

Translate this page into:
Retrospective Diagnosis of Nail-patella Syndrome
Address for correspondence: Dr. Bharat V. Shah, 35, Dr E Borges Road, Hospital Avenue, Opp Shirodkar High School, Parel, Mumbai, Maharashtra - 400012, India. E-mail: dr_bharatvshah@yahoo.co.in
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
A 37 years old female presented with asymptomatic nephrotic range proteinuria due to focal segmental glomerulosclerosis (FSGS). She was treated with steroids and mycophenolate mofetil to which there was no response and progressed to advanced chronic kidney disease. When her brother who was being evaluated as a potential donor, for renal transplant, was found to have proteinuria and a genetic study for the steroid-resistant nephrotic syndrome was done. This revealed mutation in the LMX1B gene. It is then that a diagnosis of nail-patella syndrome (NPS) was made. She underwent a successful renal transplant with her father as a donor and is doing well.
Keywords
FSGS
kidney transplant
LMX1B gene
nail-patella syndrome
nephrotic syndrome

Introduction
Nail patella syndrome (NPS) is an autosomal dominant disorder resulting from a mutation in the LMX1B gene located on the long arm of chromosome 9q34.1 and encoding the LIM-homeodomain protein LMX1B.[1] It is characterized largely by the involvement of the musculoskeletal system. Typically, it is a tetrad characterized by the involvement of the nails, elbows, knees and the presence of iliac horns. Nails may be absent, hypoplastic, or dystrophic, ridged longitudinally or horizontally, pitted, discoloured, separated into two halves by a longitudinal cleft or ridge of skin, thin or, less often, thickened.
In elbows, there may be a limitation of extension, pronation and supination, or cubitus valgus. Typical radiological findings include dysplasia of the radial head, hypoplasia of the lateral epicondyle and capitulum, and prominence of the medial epicondyle. These abnormalities may result in dislocation of the radial head usually posteriorly. Knee involvement may be in the form of small, irregularly shaped, or absent and patella and involvement may be asymmetrical.
Iliac horns are bilateral, conical, bony processes that project posteriorly and laterally from the central part of the iliac bones of the pelvis and are considered pathognomonic of NPS.
The involvement of other body systems such as the kidneys and eyes are also well documented.[123] LMX1B is highly expressed in podocytes and renal involvement occurs in 30%–50% of individuals. Nephrotic syndrome is exceptional and end-stage renal disease (ESRD) occurs in approximately 5%.[4]
It is not uncommon for families to remain undiagnosed for several generations despite having been seen by doctors from a variety of disciplines because the clinical features vary in both frequency and severity and there is inter and intrafamilial variability.[3] We report a case that initially presented with nephrotic range proteinuria due to FSGS. It was only when the genetic analysis was done that NPS was suspected and confirmed on clinical and radiological evaluation.
Case Report
A 37-year-old female, the ophthalmologist by profession, presented to us in November 2017 with advanced kidney disease.
Her complaints date back to October 2012 when she presented to her primary physician with irregular menstrual cycles. On investigations then she was found to have nephrotic range proteinuria (7650 mg/24 h) with a bland urine sediment and normal serum creatinine. She was initially started on empirical steroids. She developed diarrhoea, abdominal pain and nausea following which her steroids were withheld and a native kidney biopsy was done. On light microscopy, there were 20 glomeruli of which 6 were globally sclerosed, 2 showed segmental sclerosis with corresponding adhesion to Bowmanʼs capsule. Twelve glomeruli showed mild focal increase in mesangial matrix. There was mild tubulointerstitial fibrosis. Immunofluorescence microscopy was negative. Electron microscopy showed diffuse effacement of podocyte foot processes. Capillary loop BM was uniform and of normal thickness. There was segmental capillary loop obliteration with accumulation of lipid, hyaline and foam cells associated with capsular adhesion. Overlying epithelial cell hyperplasia was present. There was no capillary loop immune complex-like dense deposits present.
The mesangial matrix was minimally expanded without hypercellularity or electron dense deposits. The biopsy was reported as focal and segmental glomerulosclesosis (FSGS). She was again started on steroids at 1 mg/kg/day and mycophenolate mofetil (MMF) 500 mg twice a day was added. However, there was no response and in December 2014 all immunosuppressive drugs were stopped. Over the next 3 years there was progressive rise in creatinine and when she presented to us in November 2017 the eGFR was 10 mL/min and kidneys were small and echogenic on ultrasound. She was advised to undergo pre-emptive transplant.
Her brother came forward as potential donor. Initial evaluation revealed that he too had proteinuria. In view of this, genetic analysis for steroid resistant nephrotic syndrome was carried out. This revealed a mutation in exon 2 of the LMX1B gene. No other mutations were reported. Mutations in the LMX1B gene are known to be associated with the Nail-patella syndrome. This made us look for features of nail-patella syndrome in our patient. Indeed, she had nail abnormalities in the form of underdeveloped, split, ridged and pitted nails in the upper limb. Her skin was loose and wrinkled. Her patella was small and irregularly shaped on the left and absent on the right. Pelvic X-ray showed bilateral iliac horns [Figure 1].
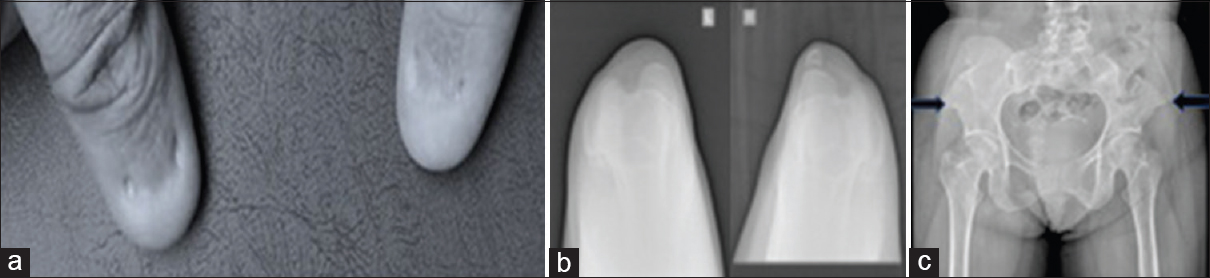
- Clinical and radiological features of nail patella syndrome a) Dysplastic nails, b) small left patella and absent right patella, c) bilateral iliac horns (arrows)
The patient's 60 years old father was next evaluated as potential donor. He had nail features like that of the patient but no renal abnormalities. The routine urinalysis was normal, urine spot protein to creatinine ratio was 0.15 and GFR was 75 mL/min. He was accepted as a donor and transplant was performed on March 13, 2018 with basiliximab as induction agent and standard triple drug maintenance immunosuppression comprising of tacrolimus, mycophenolate sodium and tapering doses of steroids. She is now 2 years posttransplant with no episode of rejection and stable allograft function with most recent creatinine being 0.77 mg/dL.
Discussion
This case highlights how Nail patella syndrome (NPS), also known as hereditary osteoonychodysplasia (HOOD), or Fong disease, can be completely missed. The syndrome is an autosomal dominant disorder characterized by abnormalities of the nails, elbows, knees and pelvis. These features are very subtle, and it is very common for families to remain undiagnosed for several generations despite having been seen by doctors from a variety of disciplines.[3] This is exactly what happened in our patient.
Our patient first presented to her physician with irregular menstrual cycles and on routine laboratory evaluation was found to have proteinuria which on quantification was found to be in nephrotic range. She was empirically treated with steroids and when she did not respond, a kidney biopsy was performed. Even on biopsy which included light microscopy, immunofluorescence microscopy and electron microscopy, NPS was not suspected. The characteristic EM lesion reported in literature in cases of NPS is a change in the glomerular basement membrane (GBM), consisting of presence of mottled and lucent rarefactions of the GBM. These changes were not seen in our patient's biopsy. We could not find any report in literature of absence of characteristic EM changes in cases of NPS with renal involvement. It is known that pathologic findings are largely unrelated to the clinical severity or prognosis.
She was treated as primary FSGS with steroids and MMF to which she had no response and she progressed to ESRD. It was only when the patient's brother was evaluated as potential donor and found to have proteinuria that a genetic aetiology of nephrotic syndrome was suspected. The genetic study revealed mutation in exon 2 of LMX1B gene which is known to be associated with NPS.[135] It is at this stage that we looked for and observed all the features of NPS in our patient.
In this case, we did genetic analysis because the brother who was being evaluated as a potential donor was found to have proteinuria. But we now feel that genetic analysis should be done in all cases of steroid resistant nephrotic syndrome. If there is a genetic cause, there may be no benefit of additional immunosuppression. Further, if the kidney disease progresses to ESRD, there would be no risk of recurrence in the allograft. Before the diagnosis of NPS was made, we informed the patient about a 30–50% risk of recurrence of FSGS after transplant.[6] However, once we received the report of genetic analysis, we explained that there was no risk of recurrence.
Clinical manifestations of NPS are extremely variable in both frequency and severity and there is inter and intrafamilial variability. This means that while clinical renal involvement may be present in one family member, it may not be present in another family member.[5] In our case, too, the patient's father had dysplastic nails (presumably the children inherited the disease from their father) but had no renal involvement as suggested by normal urinalysis, ultrasound and normal GFR. He was thus accepted as donor. The patient underwent transplant on March 13, 2018 and has had an entirely uneventful course with normal urinalysis and normal creatinine (0.77 mg/dL) when last seen on March 7, 2020. Patients with nail patella syndrome do well after kidney transplant and it should be the renal replacement modality of choice if they develop end stage kidney disease.[78]
In summary, there were many lessons to be learnt from this unusual case: (1) The patient presented with asymptomatic proteinuria which was in nephrotic range, an uncommon feature of NPS; (2) The patient progressed to ESRD which happens in only about 5% of cases of NPS; (3) The diagnosis of NPS was made only when genetic analysis showed mutation in LXMB1 gene; (4) Clinical manifestations varied within the family. Thus, the father who also had NPS did not have renal involvement and could donate kidney to his daughter.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Nail-patella syndrome. Overview on clinical and molecular findings. Pediatr Nephrol. 2002;17:703-12.
- [Google Scholar]
- Hereditary onycho-osteodysplasia (Nail-Patella syndrome). A report of nine kindreds. J Bone Joint Surg Am. 1969;51:505-16.
- [Google Scholar]
- Nail patella syndrome: A review of the phenotype aided by developmental biology. J Med Genet. 2003;40:153-62.
- [Google Scholar]
- Steroid-resistant nephrotic syndrome as the initial presentation of nail-patella syndrome: A case of a de novo LMX1B mutation. BMC Nephrol. 2017;18:100. doi: 10.1186/s12882-017-0516-7
- [Google Scholar]
- Nail-patella syndrome: Identification of mutations in the LMX1B gene in Dutch families. J Am Soc Nephrol. 2000;11:1762-6.
- [Google Scholar]
- Recurrent glomerulonephritis after kidney transplantation. Am J Transplant. 2006;6:2535-42.
- [Google Scholar]
- Living-related renal transplantation in a patient with nail-patella syndrome. Nephron. 1988;50:164-6.
- [Google Scholar]




