

Translate this page into:
Senior-Loken Syndrome: A Rare Cause of End Stage Kidney Disease in a Child

Corresponding author: Abhishek Abhinay, Division of Pediatric Nephrology, IMS, Banaras Hindu University (BHU), Varanasi, Uttar Pradesh, India. E-mail: abhinay.abhishek@bhu.ac.in
-
Received: ,
Accepted: ,
How to cite this article: Singh P, Abhinay A, Singh A, Prasad R. Senior-Loken Syndrome: A Rare Cause of End Stage Kidney Disease in a Child. Indian J Nephrol. doi: 10.25259/IJN_168_2025
Dear Editor,
A 14-year-old boy, second-born from a third-degree consanguineous marriage, presented with joint pain in both lower limbs for 2 years. Examination showed pallor, no dysmorphic features, and height and weight <3rd percentile. Oculomotor apraxia was present. Investigations revealed Hb: 5.0 g/dL, serum Ca/PO4: 6.3/8.7 mg/dL, Alkaline Phosphatase (ALP): 2194U/L, intact parathyroid hormone (iPTH): 1207.2 ng/L, 25(OH)Vit D: 128.64 ng/mL, pH: 7.30, HCO3: 18 mEq/L, and serum urea/creatinine: 256/6.6 mg/dL. Ultrasound kidney ureter bladder showed bilateral shrunken kidney. Fundus examination showed retinitis pigmentosa [Figure 1]. Senior-Loken syndrome (SLSN) due to chronic kidney disease and retinitis pigmentosa (R) was suspected. Whole exome sequencing showed IQCB1 (ENST00000310864.11): c.488-1G>A, homozygous, pathogenic, autosomal recessive variant (as per Human Genome Variation Society Classification) associated with Senior-Loken syndrome 5 (OMIM#609254), confirming the diagnosis. The child was administered maintenance hemodialysis, antihypertensive drugs, oral sodium bicarbonate, calcium, sevelamer, and calcitriol. Family genetic testing was not done due to financial constraints [Supplementary File].
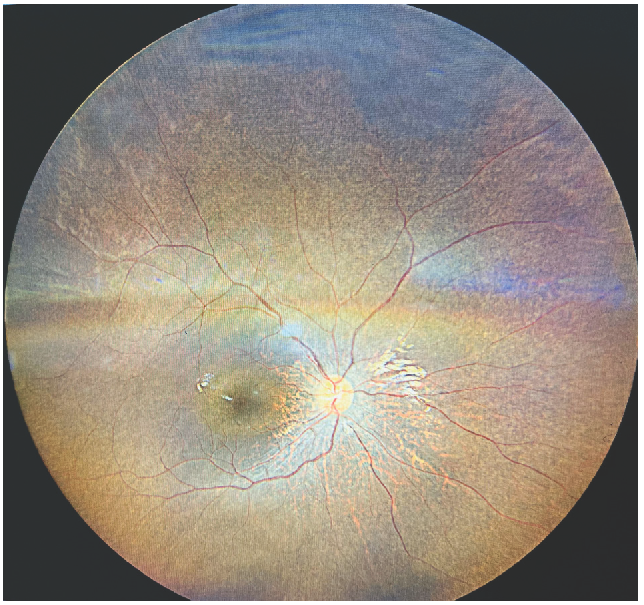
- Fundus examination (direct ophthalmoscopy) showing retinitis pigmentosa.
SLSN is a rare autosomal recessive oculo-renal disease with a prevalence of 1:1,000,000.1,2 The SLSN5 is caused by mutations in the NPHP5/ICQB1 gene, first identified by Otto et al.1 in 2005. It was first concurrently described by Senior et al.3 and Loken et al.4 in 1961 as a combination of familial juvenile nephronophthisis and Leber congenital amaurosis. Few cases have emerged from India, of which only one has been genetically proven.5 Severe infantile, juvenile, and adolescent are the three clinical nephronophthisis types. Severe infantile is the most severe form, and juvenile is the most common. The age of onset is ∼5 years, as in this case. This letter intends to raise awareness that ocular involvement is the most common presenting complaint in this syndrome. Our patient suffered from difficulty in vision since 5 years of age and was misdiagnosed with myopia and prescribed spectacles.
Conflicts of interest
There are no conflicts of interest.
References
- Nephrocystin-5, a ciliary IQ domain protein, is mutated in senior-loken syndrome and interacts with RPGR and calmodulin. Nat Genet. 2005;37:282-8.
- [CrossRef] [PubMed] [Google Scholar]
- Nephronophthisis: Disease mechanisms of a ciliopathy. J Am Soc Nephrol. 2009;20:23-35.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Juvenile familial nephropathy with tapetoretinal degeneration A new oculorenal dystrophy. Am J Ophthalmol. 1961;52:625-33.
- [CrossRef] [PubMed] [Google Scholar]
- Hereditary renal dysplasia and blindness. Acta Paediatr (Stockh). 1961;50:177-84.
- [CrossRef] [PubMed] [Google Scholar]




