

Translate this page into:
Spectrum of IgG4-related Kidney Disease at a Tertiary Care Center
Address for correspondence: Dr. M. Rathi, Department of Nephrology, Postgraduate Institute of Medical Education and Research, Chandigarh - 160 012, India. E-mail: drmanishrathi2000@yahoo.co.in
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
IgG4-related kidney disease (IgG4 RKD) is increasingly reported with varied manifestations. The present study was carried out to study the spectrum of IgG4 RKD. All patients with renal manifestation associated with conditions known to be associated with IgG4-related diseases (IgG4 RDs), or renal imaging or histology suggestive of IgG4 RKD were included and evaluated further. Patients with known extrarenal IgG4RD were also screened for renal involvement. Out of 40 patients screened over a period of 15 months, IgG4 RKD was diagnosed in 8. Majority were male (87.5%) with mean age being 56 years. Disease spectrum ranged from normal renal function in one to renal failure requiring dialysis in another two patients. Significant proteinuria was uncommon (12.5%) while hematuria was not seen in any patient. Tubulointerstitial nephritis was seen in all four patients who underwent kidney biopsy. Two patients had associated glomerular lesions in the form of immune complex crescentic glomerulonephritis. The most common imaging abnormality was hypodense renal lesions seen in 2 patients. Elevated IgG4 levels had 87.5% sensitivity and 78.3% specificity for IgG4 RKD and levels did not correlate with disease severity. Of 4 patients treated with steroids, 3 showed improvement in renal function. IgG4 RKD is an uncommon disease even at a referral tertiary care center. Elevated IgG4 levels alone are neither sensitive nor specific for the diagnosis of IgG4 RKD, and a combination of clinical, imaging, serological, and histological features are required for diagnosis.
Keywords
IgG4-related disease
IgG4-related kidney disease
tubulointerstitial nephritis

Introduction
IgG4-related disease (IgG4 RD) has been described as a chronic fibro-inflammatory condition that affects multiple organ systems and is characterized by unique histopathological features. Initially being described with respect to the pancreas as autoimmune pancreatitis Type 1, the spectrum of IgG4 RD now includes a variety of fibro-inflammatory conditions such as Riedel's thyroiditis, Mikulicz's syndrome, and Küttner's tumor that were previously considered to be single organ diseases.[1] IgG4-related kidney disease (IgG4 RKD) is a term for kidney involvement in association with IgG4 RD. It may present as acute or chronic progressive renal dysfunction. Most common presentation of IgG4 RKD is plasma cell-rich tubulointerstitial nephritis (TIN). Among the glomerular lesions, membranous glomerulopathy (MGN) is the usually observed pattern. Apart from this, it may also present as sclerosing pyelitis, ureteral inflammatory pseudotumor, renal masses, and obstructive uropathy secondary to retroperitoneal fibrosis (RPF).[2]
With the increasing awareness of IgG4 RD among clinicians and pathologists, there has been an increase in the number of reported cases. A definite diagnostic criterion has been laid down for this new disease entity by the Japanese Ministry of Health, Labour and Welfare.[3] Apart from the diagnostic criteria, serum IgG4 levels and serum IgG4/IgG ratio have also been found to have good diagnostic sensitivity and specificity.[45] However, the pathogenesis and exact etiology of this disease still remain unclear. Glucocorticoids have been found to improve IgG4-related organ dysfunction, but disease often relapses when the doses are tapered.
This study aims to find the spectrum of IgG4 RKD at a tertiary care center by screening all patients of renal impairment with clinical, histopathological, and imaging findings compatible with known presentations of IgG4 RD and by screening the patients with extrarenal IgG4 RD for renal involvement.
Methods
In this observational prospective cohort study, all patients presenting in the department of nephrology or seen in other departments as nephrology consultation, between July 2014 and September 2015, were screened for IgG4 RKD in case of unexplained acute kidney injury or acute kidney disease, unexplained urinary abnormalities, renal imaging, or histology suggestive of IgG4 RKD or if they had renal involvement with conditions known to be associated with IgG4 RD. Patients with known extrarenal IgG4 RD were also screened for renal involvement. The study was approved by the institute's ethics committee (reference number: NK/1662/DM/10369-70). Patients who had any definite etiology of renal impairment or had a history of steroid intake within the past 3 months or those who did not give consent were excluded from the study.
After informed consent, all patients underwent investigations including complete hemogram, liver function test, renal function test, urine routine and microscopy, 24 h urine protein estimation, antinuclear antibody, antineutrophil cytoplasmic antibody, serum IgG4, and total IgG levels. Ultrasonography kidney, ureter, and bladder was done in every patient, and if required, computed tomography (with or without contrast) was also done. Kidney biopsy was done where indicated and was assessed under light microscope, direct immunofluorescence, and electron microscope. The biopsy was looked specifically for features suggestive of IgG4 RKD such as presence of TIN, degree of lymphoplasmacytic infiltration, eosinophilic infiltration as well as for presence of associated glomerulonephritis, degree of fibrosis, and immune complex deposition in tubular basement membrane (TBM). If the light microscopy findings were consistent with IgG4 RKD, IgG4 staining was done and a number of positive plasma cells/hpf were counted.
The diagnosis of IgG4 RKD was made according to the Japanese Society of Nephrology criteria[6] for IgG4 RKD [Table 1]. Patients diagnosed with IgG4 RKD were treated with prednisolone prescribed at a dose of 0.6 mg/kg/day for 2–4 weeks and was tapered over a period of 3–6 months to 5.0 mg/day following which it was planned to be continued at a dose between 2.5 and 5.0 mg/day for up to 3 years.[7] Response was assessed after 3 months of treatment and was categorised as “treatment response” (reduction in 24 h urine protein >50% and/or stabilization of serum creatinine, i.e. ± 25%) or “remission” (24 h urine protein <500 mg and/or normalization of serum creatinine) or “refractory disease” (patients who did not fulfill the criteria for response or remission).

Patients were followed at week 1, 2, 4, and then, monthly for 3 months. At each follow-up, serum creatinine, urine routine and microscopy, and 24 h urine protein estimation were done. Serum IgG and IgG4 levels were done at the end of 3 months in patients treated with steroids.
Results
A total of 40 patients were screened for suspected IgG4 RKD. The reasons for screening are shown in Figure 1. Twenty-two patients had elevated IgG4 levels; of these, 17 patients had IgG4 RD (8 renal and 9 extrarenal) while the other 5 patients did not have any other evidence of IgG4 RD, and alternative diagnosis was established in each case (non-IgG4 pancreatitis in 3 patients, prostate carcinoma in 1 patient, and disseminated tuberculosis in 1 patient). Figure 2 shows flowchart of patients in the study.

- Reasons for screening

- Flow of patients in the study
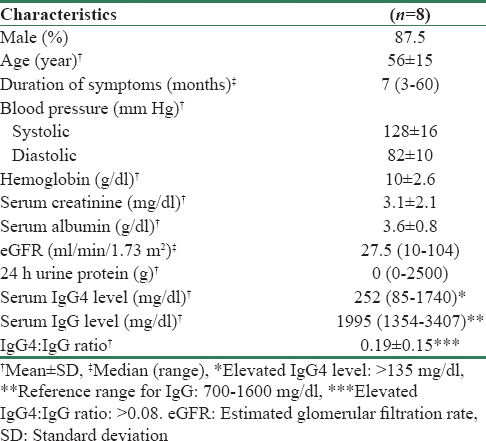
Out of the eight patients with IgG4 RKD, 3 patients had definite, 1 had probable, and 4 had possible IgG4 RKD. There were 7 males and 1 female with a mean age of 56 ± 15 years. The median duration of symptoms was 7 months. Two patients had presented as acute kidney disease while six were known kidney disease for >3 months suggesting chronic kidney disease. The most common presentation was constitutional symptoms in the form of fever and decreased appetite seen in 5 patients. Other symptoms were swelling (n = 1), nocturia (n = 3), and stress incontinence (n = 1). Four patients were hypertensive. Three patients (one in definite group and two in possible group) had been diagnosed hypertension previously while in 1 patient; hypertension was detected simultaneously with kidney disease. Three patients had evidence of extrarenal IgG4 RD (one had IgG4-related pancreatitis and two had IgG4-related RPF). Renal histological diagnosis could not be established in these three patients as one had multiple renal cysts, one patient with RPF had parenchymal thinning, while the other one with RPF did not give consent for kidney biopsy. A summary of the baseline characteristics and clinicopathological features of these 8 patients is shown in Tables 2 and 3, respectively. Only four patients underwent kidney biopsy; three were diagnosed as definite IgG4 RKD and one as probable IgG4 RKD. For patients 1 and 7, biopsy was done from the hypodense renal lesions while patients 6 and 8 underwent routine kidney biopsy. There was no correlation between serum IgG4 levels and number of IgG4 positive plasma cells in kidney biopsy. In fact, the patient with maximum number of IgG4 plasma cells in biopsy had the lowest serum IgG4 levels. Kidney biopsy findings are summarised in Table 4.
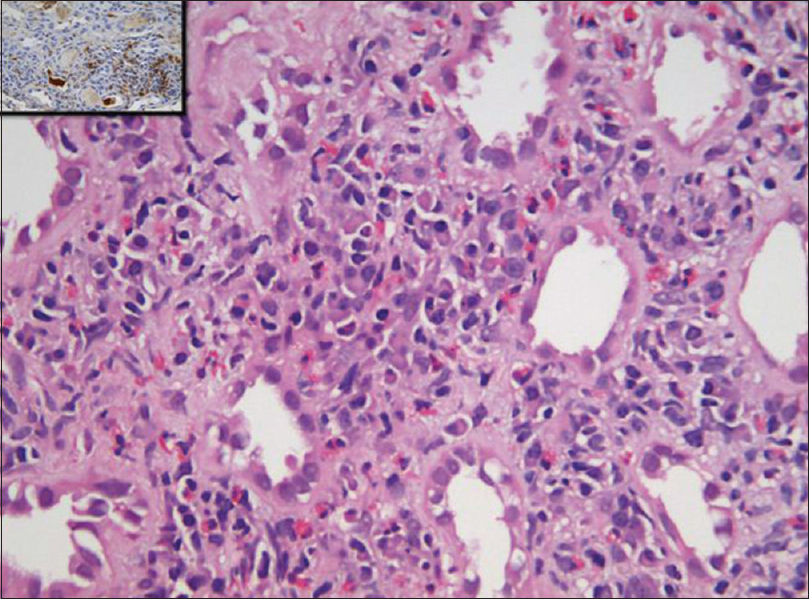
- Photomicrograph of kidney biopsy shows inflammation composed of lymphoplasmacytic cells along with eosinophils in insinuating pattern. Plasma cells were positive for IgG4 immunostain (inset) (H and E, IHC IgG4 inset: ×40)

- Photomicrograph of kidney biopsy shows (a) membranoproliferative pattern with cellular crescent and plasma cell-rich tubulointerstitial nephritis, (b) IgG4-positive plasma cells in interstitium and IgG4 positivity in immune complexes along glomerular capillaries, (c and d) show IgG and c3 positivity in glomerular capillaries and mesangium (a: H and E, ×20, b: IHC for IgG4 ×40, c: FITC IgG (×20), d: FITC C3 (×20). (IgG4 RD: IgG4-related disease, IgG4 RKD: IgG4-related kidney disease)


Out of 5 patients screened for imaging suggestive of IgG4 RKD, 3 were diagnosed with IgG4 RKD (2 definite, 1 probable). Out of 6 patients screened for suggestive histology, 1 was diagnosed with definite IgG4 RKD. Out of 13 patients screened because of renal involvement with conditions known to be associated with IgG4 RD (unexplained pancreatitis: n = 6, primary biliary cirrhosis: n = 1, bulky kidneys: n = 1, RPF: n = 2, aortic aneurysm: n = 1, parotid enlargement: n = 1, and bulky prostate: n = 1), 1 (with aortic aneurysm) was diagnosed with possible IgG4 RKD. Out of 13 patients with extrarenal IgG4 RD (IgG4 pancreatitis: n = 9, IgG4 RPF: n = 3, and IgG4 esophagitis: n = 1), 3 were diagnosed with possible IgG4 RKD.
A total of four patients were treated with steroids. These included the three definite cases and one possible case. Repeat IgG4 and IgG levels were done in three (2 definite, 1 possible) of these patients who had completed 3 months of treatment. There was decrease in both the IgG and IgG4 levels after 3 months of therapy. Furthermore, serum creatinine showed improving trend in all three patients but did not normalize in any of them indicating response to therapy but not remission [Table 5]. One of these patients who had presented with dialysis-dependent renal failure had marked decrease in serum creatinine with steroids and had become dialysis free after 1 month of therapy. There was no proteinuria in all the three patients at baseline. The fourth patient had presented with dialysis-dependent renal failure and had crescents on kidney biopsy. Thus, he was treated with three methylprednisolone pulses followed by 1 mg/kg prednisolone. However, he did not show any response and died in the 3rd month of therapy due to inadequate dialysis. Of the four patients who did not undergo therapy, all had stable renal function on follow-up.

Analyzing the 31 patients with renal abnormality (on urinalysis, imaging, histology, or elevated creatinine levels), IgG4 levels were elevated in 7 of 8 patients with IgG4 RKD and in 5 of 23 patients without IgG4 RKD, giving IgG4 levels a sensitivity and specificity of 87.5% and 78.3%, respectively, for the diagnosis of IgG4 RKD.
Discussion
A total of 40 patients were screened for IgG4 RKD over a period of 15 months, of which 22 patients had elevated IgG4 levels, and of these, 17 had IgG4 RD. Among these 17 patients, 9 had only extrarenal IgG4 RD and 8 had IgG4 RKD (3 definite, 1 probable, and 4 possible).
Majority of the patients with IgG4 RKD were male (n = 7; 87.5%) with mean age at presentation being 56 ± 15 years. Previous studies have also found a male preponderance, but mean age varied from 47 to >60 years.[8910] However, there were individuals as young as 20–30 years also in those studies. In the present study, 7 patients had renal dysfunction at presentation with mean serum creatinine of 3.1 mg/dl. This is similar to previous studies where up to 77% of patients had renal dysfunction at presentation.[1011] Proteinuria >1 g/day was seen only in 1 (12.5%) patient; this is also similar to previous studies which have reported significant proteinuria in 9%[11] to 30%[10] of patients which is in concordance with the tubulointerstitial nature of the disease. None of the patients in the present study had hematuria. As such, hematuria is not a common feature of tubulointerstitial diseases and has been reported in only 9%–22% of cases in previous series of IgG4 RKD.[1011]
Of the total 40 patients, 6 were screened as they had histological findings of TIN suggestive of IgG4 RKD (i.e plasma cell rich tubulointerstital nephritis); however, after staining for IgG4 only 1 (17%) out of these 6 was finally diagnosed as IgG4 RKD, while others were diagnosed to have non IgG4 related acute interstitial nephritis Similar were the results of the study by Nada et al.,[8] where only 37% of patients with TIN suggestive of IgG4 RKD on light microscopy actually qualified for IgG4 TIN. This implies that light microscopic findings could be deceptive in cases with TIN unless the presence of IgG4+ plasma cells has been demonstrated by immunohistochemistry and other features including imaging and serology are also supportive. All the patients with IgG4 RKD who underwent kidney biopsy in the present study had TIN, lymphoplasmacytic infiltration, increased IgG4-positive plasma cells (>10 IgG4+ plasma cells/hpf), and insinuating storiform fibrosis as seen in previous studies.[6101112] TBM deposits have been reported in 10%–80% of patients[11] and have been suggested to correlate with disease progression. However, none of our patients had TBM deposits, and even in the previous studies, the precise significance of these TBM deposits seems unclear as there is no consistency in their composition, and various immunoglobulin and complement proteins including IgG, IgA, C1q, and C3 have been observed in these deposits.[12]
Although MGN is the most common glomerular disease associated with IgG4 RKD, a number of other glomerular diseases have also been reported in IgG4 RKD. These include endocapillary proliferative nephritis, crescentic glomerulonephritis, IgA nephropathy, Henoch-Schonlein purpura nephritis, and membranoproliferative glomerulonephritis.[131415] Two out of four of our patients who underwent kidney biopsy had immune complex crescentic GN. Imaging abnormalities were identified in three patients in the form of hypodense renal lesions (n = 2) and bulky kidneys (n = 1). This is similar to previous studies where patchy hypodense lesions were the most common imaging abnormalities, and it was seen that radiological lesions do not correlate with renal function.[1011] Two patients with very high IgG4 levels had only mild renal dysfunction, while one patient with severe renal dysfunction had relatively low levels of IgG4. It has been reported earlier that 10% of patients with IgG4 RKD may not have elevated IgG4 levels and that IgG4 levels do not correlate with severity of renal disease.[810] Furthermore, there was no correlation between the serum IgG4 levels and the radiological findings.
Three out of four patients in the present study who received steroids completed 3 months of follow-up and all three of them responded to treatment including the one who was dialysis dependent at presentation. It has been seen previously that even patients with extensive fibrosis showed improvement while those with minimal chronic changes have also progressed to end-stage renal disease despite treatment.[8101112] Another finding noticed in our as well as previous studies was that many untreated patients had either stable serum creatinine or mild increase in serum creatinine over long-term follow-up.[10] These observations raise the doubt that whether steroids or immunosuppression actually plays a significant role in modifying the natural course of IgG4 RKD.
Of the 22 patients who had elevated serum IgG4 levels, 5 (23%) had diagnosis other than IgG4 RD. Previous studies have also shown that elevated serum IgG4 concentration is not specific for IgG4 RD and may be seen in variety of other diseases.[51617] In a retrospective analysis from Mayo clinic, only 18.4% of patients with elevated IgG4 levels actually had definite or probable IgG4 RD[17] suggesting that IgG4 levels alone are unreliable for the diagnosis of IgG4 RD. In fact, it has been seen that up to 5% of the normal healthy population may have elevated IgG4 levels.[18] From the available data, it seems that IgG4 is not pathognomonic of IgG4 RD and it is not clear whether IgG4 has a direct role in the pathogenesis of IgG4 RD or is just a marker of the disease.
Given the nonspecific nature of serum IgG4 levels, ordering it for every patient with unexplained renal dysfunction is not justified. Rather, importance should be given to constellation of presenting symptoms, laboratory findings, imaging characteristics, and histology to accurately diagnose IgG4 RKD.
Conclusion
IgG4 RKD is an uncommon disease even at a tertiary referral institute. Patients usually have nonspecific constitutional symptoms, and a high index of suspicion is required to diagnose IgG4 RKD.
Financial support and sponsorship
This study was financially supported by the Indian Council of Medical Research.
Conflicts of interest
There are no conflicts of interest.
References
- Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. 2012;22:21-30.
- [Google Scholar]
- Cutoff values of serum IgG4 and histopathological IgG4+plasma cells for diagnosis of patients with IgG4-related disease. Int J Rheumatol. 2012;2012:580814.
- [Google Scholar]
- The diagnostic utility of serum IgG4 concentrations in IgG4-related disease. Ann Rheum Dis. 2015;74:14-8.
- [Google Scholar]
- Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011;15:615-26.
- [Google Scholar]
- IgG4-related tubulointerstitial nephritis: A prospective analysis. Int J Rheum Dis. 2016;19:721-9.
- [Google Scholar]
- The clinical course of patients with IgG4-related kidney disease. Kidney Int. 2013;84:826-33.
- [Google Scholar]
- Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol. 2011;22:1343-52.
- [Google Scholar]
- Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int. 2010;78:1016-23.
- [Google Scholar]
- Immunohistochemical characteristics of IgG4-related tubulointerstitial nephritis: Detailed analysis of 20 Japanese cases. Int J Rheumatol. 2012;2012:609795.
- [Google Scholar]
- Membranoproliferative glomerulonephritis-like glomerular disease and concurrent tubulointerstitial nephritis complicating IgG4-related autoimmune pancreatitis. Intern Med. 2009;48:157-62.
- [Google Scholar]
- Endocapillary proliferative glomerulonephritis with crescent formation and concurrent tubulointerstitial nephritis complicating retroperitoneal fibrosis with a high serum level of IgG4. Clin Nephrol. 2007;68:308-14.
- [Google Scholar]
- A case of IgG4-related tubulointerstitial nephritis concurrent with Henoch-Schönlein purpura nephritis. Allergy Asthma Clin Immunol. 2011;7:5.
- [Google Scholar]
- Detection of serum IgG4 levels in patients with IgG4-related disease and other disorders. PLoS One. 2015;10:e0124233.
- [Google Scholar]
- Spectrum of disorders associated with elevated serum IgG4 levels encountered in clinical practice. Int J Rheumatol. 2012;2012:232960.
- [Google Scholar]
- Serologic issues in IgG4-related systemic disease and autoimmune pancreatitis. Curr Opin Rheumatol. 2011;23:108-13.
- [Google Scholar]




