

Translate this page into:
Successful Kidney Transplantation in a Young Male with Type 2 Xanthinuria
Corresponding author: Rajasekara Chakravarthi Madarasu, HOD & Clinical Director, Department of Nephrology, Yashoda Hospital, Hitech City, Hyderabad, Telangana, India. E-mail: rajasekarac@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Kota M, Madarasu RC, Penmetsa VV, Gutta S, Naidu A. Successful Kidney Transplantation in a Young Male with Type 2 Xanthinuria. Indian J Nephrol. 2024;34:403-5. doi: 10.25259/ijn_509_23
Abstract
Type-II Xanthanuria is an genetic disorder associated with diminished serum uric acid levels. Patients with xanthanuria has absence of xanthine oxidase or xanthine dehydrogenase activity, the enzyme that converts hypoxanthine to xanthine and xanthine to uric acid. Deficiency of these enzyme leads to elevated levels of xanthine in urine which further leads to precipitation of xanthine in urine which further helps to formation of renal stones and ultimately leads to chronic kidney disease and end stage renal disease. We report a 23 years old male, who reached ESRD due to Type 2 xanthinuria, which was confirmed by genetic studies, who later successfully underwent renal transplant surgery and currently having normal life with functioning graft.
Keywords
End-stage renal disease
Renal transplantation
Xanthinuria
Uric acid
Urolithiasis

Introduction
Xanthinuria type - II, a rare hereditary disease is characterized by a marked and isolated deficiency of xanthine dehydrogenase activity in the tissues, usually in xanthinuric. Clinically this leads to formation of xanthine stones in kidney which further leads to ESRD secondary xanthine stones in kidney. The following cases highlight the importance of evaluating renal calculus causes in young males, conducting renal transplant workups for xanthinuria patients, and managing post-transplant care.
Case Report
A 23-year-old male was incidentally found to have elevated serum creatinine of 24 mg/dL with bilateral renal stones and metabolic acidosis in 2011. He was on hemodialysis for a couple of weeks after which the kidney function recovered and he came off dialysis. Two years later, he underwent extracorporeal shock wave lithotripsy (ESWL) and bilateral double J (DJ) stenting. However he prgressed to ESRD by 2017 when he was initiated on maintenance hemodialysis. As he had oxaluria, elevated blood oxalate, with a history of third-degree consanguineous marriage parents, primary hyperoxaluria was suspected and whole genome sequencing (WGS) was done. WGS revealed mutation in MOCOS gene, which encodes molybdenum cofactor sulfurase, necessary for the normal function of xanthine dehydrogenase and aldehyde oxidase. A diagnosis of type 2 Xanthinuria was made. He also had very low serum uric acid level (0.02 mg/dL). In June 2021, he was taken up for kidney transplantation surgery with his mother as donor. Transplant surgery was uneventful, currently his serum creatinine is 0.9 mg/dL (as on 20/05/2023), serum uric acid is 0.5 mg/dL, and he is on triple immunosuppression. All necessary steps to prevent formation of Xanthine stones in graft kidney like maintaining good hydration, providing oral potassium citrate, following serum uric acid level on every fortnightly is being done.
Discussion
Hereditary xanthinuria, a rare metabolic genetic disorder, was first described in 1954 by Dent and Philpot.1 It is characterized by an absence of xanthine oxidase (XO) or xanthine dehydrogenase (XDH) activity,2 an enzyme that converts hypoxanthine to xanthine and xanthine to uric acid. Enzyme deficiency leads to elevated xanthine and hypoxanthine (oxypurines) in urine. Xanthinuria is transmitted as an autosomal recessive mode. It has two types: Type I is due to an XDH-gene mutation on chromosome 2p22q,3 while Type II is a result of concurrent deficiencies of XDH and aldehyde oxidase (AO). AO gene is located on chromosome 2q33 locus.4 The disease is predominant in males and can occur at any age. The disease is manifested by urinary calculi in 30%–40% of patients. The most frequent symptoms of urolithiasis in children are abdominal pain (44%),5 hematuria (38%), fever (15%), and other symptoms secondary to urinary tract infection (UTI). Our patient also presented with urolithiasis and UTI. These complications are explained by the low solubility of the xanthine causing precipitation of xanthine crystals in the kidneys and urinary tract.6
Xanthine stones are round and brown in morphology and are radiolucent. They are generally formed of pure xanthine or sometimes associated with hypoxanthine [Figure 1]. Uric acid stones are also radiolucent, so they should be considered in differential diagnosis. However, uric acid stones are associated with hyperuricosuria and xanthine stones are associated with hypouricemia and hypouricosuria.6 Our patient had both hypouricemia and hypouricosuria. Oxypurines solubility in urine is pH-dependent with xanthine supersaturation at acid pH, leading to stone formation. In our case, urine pH was in acidic range predisposing him for stone formation. The determination of XO activity or genetic testing for involved genes confirms the diagnosis. The XO activity is near zero in homozygous and around 50% in heterozygotes. In our case, the classical presentation and WGS analysis lead to the diagnosis of classical xanthinuria type II.
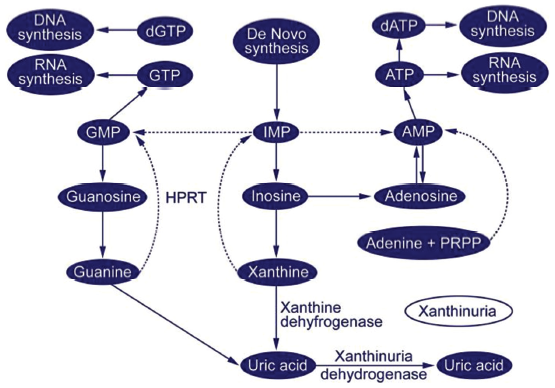
- Purine metabolic pathway (downloaded from Medscape). DNA: deoxyribonucleic acid, GTP: guanosine tri-phosphate, RNA: ribonucleic acid, GMP: guanosine monophosphate, IMP: ionosine monophosphate, AMP: adenosine monophosphate, dGTP: deoxyguanosine triphosphate, dATP: deoxy adenosine triphophate, ATP: adenosine triphosphate, PRPP: phosphoribosyl pyrophosphate, HPRT: Hypoxanthine-guanine phosphoribosyltransferase.
Management of hereditary xanthinuria helps in avoiding complications. It comprises high fluid intake (>3 l/24 h), urine alkalinization, low-purine diet avoiding vigorous exercise. The treatment modality of xanthine stones depends on their location and size. Surgical stone removal can be reserved for urinary obstruction when lithotripsy has failed to relieve it. Urolithiasis in children must be routinely evaluated for metabolic causes.
Conclusion
To our knowledge, there is no report published regarding successful renal transplant in type 2 Xanthinuria. This patient demonstrate that it is possible and have good long term outcomes after renal transplant in patients suffering from ESRD secondary to Type 2 Xanthinuria.
Conflicts of interest
There are no conflicts of interest.
References
- Asymptomatic hereditary xanthinuria: A case report. Jpn J Med. 1990;29:287-91.
- [CrossRef] [PubMed] [Google Scholar]
- Compherensive pediatric nephrology. In: Hoppe B, ed. Urolithiasis and Nephrocalsinosis in Childhood. Philadelphia: Mosby Elsevier; 2003. p. :515.
- [Google Scholar]
- Xanthinuria type I: A rare cause of urolithiasis. Pediatr Nephrol. 2007;22:310-4.
- [CrossRef] [PubMed] [Google Scholar]
- Hereditary xanthinuria type II associated with mental delay, autism, cortical renal cysts, nephrocalcinosis, osteopenia, and hair and teeth defects. J Med Genet. 2003;40:e121.
- [CrossRef] [PubMed] [Google Scholar]
- Unmeasurable uric acid in blood and urine; xanthine dehydrogenase deficiency. Rev Med Int. 1999;20:445.
- [CrossRef] [PubMed] [Google Scholar]
- Hypouricemia, an old subject and new concepts. Presse Med. 2004;33:555-6.
- [CrossRef] [PubMed] [Google Scholar]




