

Translate this page into:
Type 3 renal tubular acidosis
Address for correspondence: Dr. Rudra Goswami, Abhyudoy Housing, Flat – 18/14, ECTP, Ph – IV, Type – B, EM Bypass, Kolkata - 700 107, India. E-mail: rudra.goswami@gmail.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Renal tubular acidosis (RTA) is a group of transport defects in the reabsorption of bicarbonate, the excretion of hydrogen ion (H+), or both, resulting in systemic acidosis and hypokalemia with a normal glomerular filtration rate. Although isolated proximal (type 2) or distal (type 1) tubular pathologies are well characterized, a combined pathology leading to type 3 RTA is very rare. Here, we report a case of type 3 RTA, using an algorithmic approach to classify a scenario of hypokalemic metabolic acidosis in the setting of episodic flaccid paralysis.
Keywords
Algorithmic approach
India
renal tubular acidosis

Introduction
Renal tubular acidosis (RTA) is a group of transport defects in the reabsorption of bicarbonate (HCO3), the excretion of hydrogen ion (H+), or both, resulting in systemic acidosis and hypokalemia with a normal glomerular filtration rate.[1] This group of disorders may present with episodic weakness indistinguishable from familial or thyrotoxic periodic paralysis.[2] We present a case with emphasis on algorithmic approach using urine electrolyte excretion pattern towards reaching a diagnosis.
Case Report
A 44-year-old man presented with flaccid paralysis of all four limbs, and tetanic spasm of the hand. He had a history of a similar episode 1 year ago that progressed to respiratory paralysis, needing assisted ventilation. The past episode was attributed to hypokalemia and hypocalcemia. He received treatment outside, under an empirical diagnosis of familial periodic paralysis (FPP), with oral potassium and calcium supplementation, with acetazolamide to combat muscle weakness. The patient was stringently taking the drugs.
He was normotensive (120/80 mmHg ± 8/4 mmHg), with normal blood sugar (fasting 86 mg/dL, post prandial 112 mg/dL), and normal renal function (urea 11 mg/dL and creatinine 0.6 mg/dL). The present episode was also associated with persistent hypokalemia (K+ 1.8 (±0.45) mmol/L) and hypocalcemia [Table 1]. Arterial blood gas showed hyperchloremic metabolic acidosis (plasma HCO 3 - 12.0 mEq/L, pH 7.21, anion gap 12 mEq/L). Abdominal ultrasonography revealed bilateral nephrocalcinosis. Routine urinalysis showed glycosuria and 3+ proteinuria by dipstick.

Historically, we excluded important extrarenal causes of hypokalemia such as chronic diarrhea or laxative abuse. Documented normotension excluded aldosteronism, Cushing's syndrome, congenital adrenal hyperplasia, renovascular hypertension, and Liddle syndrome.[3]
Next, urinary electrolytes were measured with calculation of the following parameters [Table 2]:
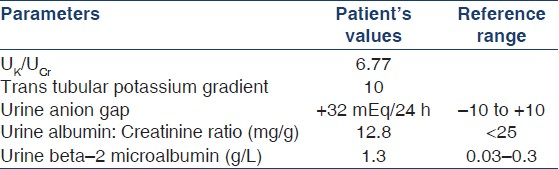
-
UK/UCr = Urine potassium (mmol/day) divided by urine creatinine (mmol/day)
-
Trans tubular potassium gradient (TTKG) = (UK/PlasmaK)/(UOsmolarity/POsmolarity)
-
Urine anion gap (UAG) = UNa + UK – UCl
The high UK/UCr (>1.5) and TTKG (>5) with positive UAG suggested renal potassium loss and acidification defect.[3–5] Urine pH was 6.7 in the face of systemic acidosis (arterial blood pH = 7.21). These along with bilateral nephrocalcinosis suggested a diagnosis of distal or type 1 RTA (dRTA). His liver (bilirubin – 0.4 mg/dL, alanine aminotransferase 23 IU/L, aspartate aminotransferase 27 IU/L) and thyroid function tests (thyroid stimulating hormone level 1.2 μIU/mL, free thyroxine level 0.9 ng/dL) were normal. Serum vitamin D level was 23.45 (normal 8-40) ng/mL and serum parathormone level was 55 (normal 10-65) pg/mL. Serum protein electrophoresis, antinuclear antibody, anti-dsDNA, anti-Ro, and anti-La; antimitochondrial antibodies; and antithyroid peroxidase and antithyroglobulin antibodies were all negative.
Next, we measured beta-2 microglobulin concentration in a 24-h urinary sample and urinary albumin to creatinine ratio (UACR). High beta-2 microglobulin and low UACR levels established proximal tubular dysfunction.[6]
To further consolidate the diagnosis, we put the patient on oral Shohl's solution (custom-made by in-house biochemistry department providing 1 mEq/mL of alkali) at a dose of 10 mEq/Kg body weight of HCO3- for 2 weeks, which restored arterial potassium level to 22 mEq/L. We then estimated fractional excretion of HCO3-, which came out to be 24%. Tubular reabsorption of phosphate was determined to be 78%. These, along with qualitative demonstration of glycosuria by Benedict's method, established a proximal tubular origin of acidosis as well.
Thus, we finally determined that the patient was suffering from combined or type 3 RTA, presumably from inappropriate acetazolamide therapy.
The patient was weaned from acetazolamide. He was put on oral Shohl's solution with potassium supplementation. Muscle weakness improved and he was finally discharged. He has been on follow-up for 6 months without any further episode of muscle weakness.
Discussion
The term RTA is applied to a group of transport defects in the reabsorption of HCO3-, the excretion of H+, or both.[1] RTA syndromes are characterized by a relatively normal glomerular filtration rate (GFR) and hyperchloremic metabolic acidosis accompanied by normal plasma anion gap.
Type 1 or dRTA is characterized by the inability to lower urinary pH to less than 5.5 in the setting of systemic acidemia. A mild degree of bicarbonaturia may be obligatorily present (<5% of the filtered load). Hypercalciuria, hypokalemia, nephrocalcinosis, and nephrolithiasis are common occurrences. Other than sporadic or hereditary causes, it may be acquired as a consequence of hypergammaglobulinemia, hyperparathyroidism, collagen vascular disorders (systemic lupus erythematosus and Sjögren syndrome), and chronic liver disease (chronic active hepatitis and primary biliary cirrhosis), and secondary to drugs or toxins.[1] Type 2 or proximal RTA may occur as an isolated entity or more commonly as a part of generalized proximal tubular dysfunction (Fanconi's syndrome).[17] It may be hereditary, or secondary to drugs (acetazolamide, topiramate, etc.) or toxins or associated with other diseases (vitamin D deficiency, hyperparathyroidism, Wilson's disease, etc.).[18] Hypokalemia is restricted to patients with Fanconi's syndrome.
In the face of symptomatic hypokalemia with acidosis, the first step toward a diagnosis is to determine the source of potassium loss or the presence of transcellular shift. High 24-h urine potassium concentration (>30 mEq/24 h) or quicker spot sampling tests (UK/UCr >1.5 and TTKG >5) indicate renal loss of potassium.[34] Next, we determined the rate of ammonium (NH4+) excretion by UAG.[9] In metabolic acidosis, the kidneys increase NH3 synthesis. The increased unmeasured NH4+ increases the measurable Cl- in urine, thereby giving a negative UAG. Conversely, a positive UAG in metabolic acidosis suggests dRTA. Also, alkaline urine during systemic acidosis and bilateral nephrocalcinosis strongly suggested dRTA. Going along this algorithmic approach and excluding secondary causes of distal tubular defects, we arrived at the diagnosis of dRTA.
As proteinuria is not a component of dRTA, we measured UACR (for glomerular proteinuria) and urine beta-2 microglobulin level (for tubular proteinuria). Isolated high beta-2 microglobulin confirmed proximal tubular dysfunction as well. Proximal RTA is characterized by a decreased renal HCO3- threshold. With low plasma HCO3- concentration, urine pH of the patients reaches below 5.5. However, when plasma HCO3- concentration is normalized, the distal nephron is overwhelmed by the large delivery of HCO3-, and fractional excretion of HCO3- increases.[8] This was demonstrated in our patient. We also demonstrated phosphaturia and glycosuria, both features of proximal tubular dysfunction. Thus, he was finally diagnosed as suffering from combined proximal and distal RTA (type 3 RTA). Specific aminoaciduria could not be demonstrated, as it was too costly for the patient.
Type 3 RTA is a rare entity. The only condition associated with it is Marble bone disease (congenital type II carbonic anhydrase deficiency).[10] There is a previous report of iatrogenic type 3 RTA due to topiramate misuse.[11] Our patient received acetazolamide due to a misdiagnosis of FPP. FPP and RTA share similar clinical characteristics, but tetany or cardiac arrhythmia suggests RTA.[2] Although acetazolamide is useful in improving muscle strength in FPP,[12] it often aggravates acidosis and hypokalemia in RTA.[2]
To the best of our knowledge, this is probably the first reported case of type 3 RTA in India. We emphasize the use of the algorithmic approach adapted here to classify and manage any patient with hypokalemic metabolic acidosis correctly.
Source of Support: No funds or grants either in cash or kinds were in any forms accepted during the clinical study
Conflict of Interest: None declared
References
- Renal tubular acidosis presenting as respiratory paralysis: Report of a case and review of literature. Neurol India. 2010;58:106-8.
- [Google Scholar]
- Diagnosis of hypokalemia: A problem-solving approach to clinical cases. Iran J Kidney Dis. 2008;2:115-22.
- [Google Scholar]
- Differentiation of glomerular, tubular, and normal proteinuria: Determinations of urinary excretion of beta-2-macroglobulin, albumin, and total protein. J Clin Invest. 1969;48:1189-98.
- [Google Scholar]
- Persistent isolated proximal renal tubular acidosis-A systemic disease with a distinct clinical entity. Pediatr Nephrol. 1994;8:70-1.
- [Google Scholar]
- Consultation with the specialist: Renal tubular acidosis. Pediatr Rev. 2001;22:277-87.
- [Google Scholar]
- The urine anion gap: A clinically useful index of ammonium excretion. Am J Med Sci. 1986;292:198-202.
- [Google Scholar]
- Topiramate induces type 3 renal tubular acidosis by inhibiting renal carbonic anhydrase. Nephrol Dial Transplant. 2006;21:2995-6.
- [Google Scholar]
- Improvement of muscle strength in familial hypokalaemic periodic paralysis with acetazolamide. J Neurol Neurosurg Psychiatry. 1988;51:1142-5.
- [Google Scholar]




