

Translate this page into:
Unusual Ultrastructural Features in a Case of C3 Glomerulopathy
Address for correspondence: Dr. Vineeta V. Batra, Department of Pathology, Academic Block, Govind Ballabh Pant Institute of Postgraduate Medical Research and Education, 2, Jawaharlal Nehru Marg, New Delhi - 110002, India. E-mail: vvbatra9@gmail.com
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Introduction
Complement 3 glomerulopathy (C3G) is characterized by glomerular deposition of complement 3 (C3) with minimal or absent immunoglobulin secondary to alternative complement pathway (AP) dysregulation.[1] The clinical picture often overlaps with that of atypical postinfectious glomerulonephritis (aPIGN); however, the long-term prognosis in both diseases is vastly different. Here, we describe one such case of a 5-year-old female child who presented with clinical features of aPIGN, pathological findings intermediate between aPIGN and C3G, and the unusual presence of tubuloreticular inclusions (TRIs) on electron microscopy (EM).
A 5-year-old female presented with repeated episodes of fever, hematuria, urinary tract infection, and generalized body edema for 2 and a half years. On clinical examination, she was normotensive, without rash, edema, or enlarged tonsils. Urinalysis showed active sediments with nephrotic-range proteinuria. Serum creatinine, complements, ASO (antistreptolysin O), and DNAse B (deoxyribonuclease B) titers were within normal limits. ANA (antinuclear antibody); dsDNA (double-stranded DNA); hepatitis A, B, C; and HIV (human immunodeficiency virus) serologies were negative. On the basis of active sediments in urine and repeated episodes of hematuria, the possibility of aPIGN was considered and percutaneous renal biopsy was performed.
The renal biopsy on light microscopy contained seven glomeruli [Figure 1a], two of which showed fibrocellular crescents [Figure 1b and d]. There was an increase in mesangial matrix and cellularity [Figure 1e] with focal endocapillary proliferation and infiltration by polymorphs. The basement membrane showed splitting on silver methenamine stain [Figure 1c]. There was mild interstitial fibrosis and tubular atrophy. Small arteries showed medial sclerosis with no evidence of active vasculitis. Direct immunofluorescence studies showed 13 glomeruli with diffuse, granular deposition of C3 (3+), immunoglobulin M (IgM; 2+), kappa (2+), and lambda (2+) along the peripheral capillary walls and mesangium [Figure 1f]. IgG, IgA, and Clq were negative. Two glomeruli showed segmental staining for fibrinogen along the Bowman's capsule, representing crescents [Figure 1g]. No immunoglobulin or complement deposits were noted in tubular basement membranes.
![(a) Light microscopy pictures of the case at low power showing well-maintained parenchyma (periodic acid Schiff [PAS] 40× magnification); and (b) enlarged glomeruli with crescents (PAS 100×); (c) glomerulus showing partial cellular crescent (arrow) with an increase in mesangial matrix and cellularity, (d) focal endocapillary proliferation (arrow) (PAS 400×); and (e) focal basement membrane splitting (Systemic Mastocytosis [SM] stain 400×); (f) Immunofluorescence (IF) figures showing granular staining of C3 in peripheral capillary walls and mesangium (200×); (g) IF figure showing deposition of fibrinogen stain around Bowman's capsule representing crescent (200×)](/content/170/2022/32/4/img/IJN-32-387-g001.png)
- (a) Light microscopy pictures of the case at low power showing well-maintained parenchyma (periodic acid Schiff [PAS] 40× magnification); and (b) enlarged glomeruli with crescents (PAS 100×); (c) glomerulus showing partial cellular crescent (arrow) with an increase in mesangial matrix and cellularity, (d) focal endocapillary proliferation (arrow) (PAS 400×); and (e) focal basement membrane splitting (Systemic Mastocytosis [SM] stain 400×); (f) Immunofluorescence (IF) figures showing granular staining of C3 in peripheral capillary walls and mesangium (200×); (g) IF figure showing deposition of fibrinogen stain around Bowman's capsule representing crescent (200×)
On EM, some glomerular basement membranes (GBM) were markedly thickened by dense osmiophilic sausage-shaped deposits [Figure 2a and b]. Others showed less dense granular deposits in the basement membrane and mesangium [Figure 2c and d]. Scattered subendothelial deposits and occasional subepithelial humps [Figure 2e and f; arrows] were identified. In addition, many TRI were identified in the cytoplasm of glomerular endothelial cells [Figure 2 g and h]. Based on the EM findings, a diagnosis of C3 glomerulopathy with features overlapping between C3 glomerulonephritis (C3GN) and dense deposit disease (DDD) with TRIs were rendered.
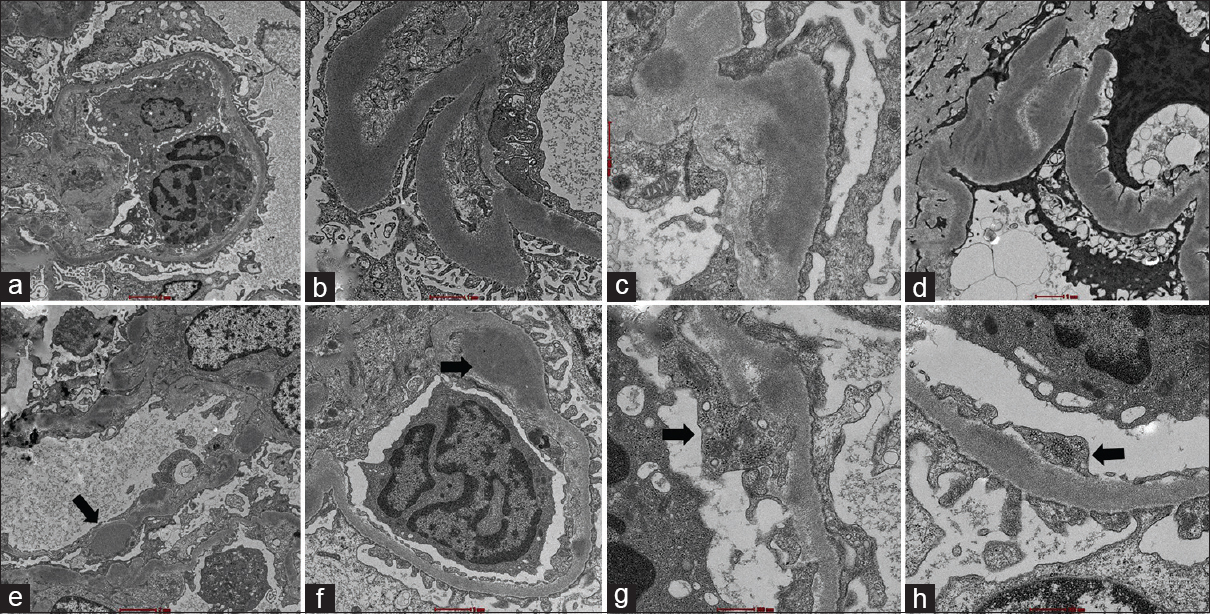
- Electron microscopy figures of the case showing two types of electron-dense deposits within basement membrane with figures (a and b) showing sausage-shaped deposits and (c and d) showing granular deposits merging with the basement membrane material. (e and f): Occasional subepithelial humps and subendothelial deposits were seen (arrows). (g, h): Numerous tubuloreticular inclusions were seen (arrows) (magnifications: a, ×2,400; b, ×6,400; c, ×6,400; d, ×3,400; e, ×2,400; f, ×3,400; g, ×6,400; h, ×6,400; respectively
The patient was discharged on steroids with no further episodes of gross hematuria being reported. On the last follow-up, 18 months after the renal biopsy, the renal function tests were within normal range. Active sediments in urine and decreased C3 levels (51 mg/dL) suggested an ongoing active disease.
Discussion
Abnormalities of the alternative complement pathway result in a spectrum of disorders, including atypical hemolytic uremic syndrome, C3G, and aPIGN.[1] aPIGN is a common cause of glomerulonephritis in children and adults that fails to resolve in 6 weeks due to persistent activation of the AP arising from defects in its regulating mechanisms.[2]
C3G is characterized by abnormalities in the AP regulators leading to continuous deposition of C3, and complement debris occurs in the GBM with attendant glomerular injury and a proliferative response.[1] The diagnosis of C3G is confirmed on EM, where it is further classified into C3GN and DDD.[3] DDD is characterized by dense osmiophilic “sausage-shaped” deposits in the GBM with a “Chinese calligraphy-like” appearance.[4] C3GN is characterized by granular subendothelial and mesangial electron-dense deposits. Ill-defined subendothelial, intramembranous, and subepithelial humpy deposits may also be present.
Cases may present with an overlap of EM findings as was seen in our case. This can be explained by their common etiology. The proteomic profile of C3GN and DDD also shows significant overlap in the composition of the GBM deposits.[5] There is also considerable overlap between the clinical and histopathological features between C3G and aPIGN. C3GN cases are often preceded by upper respiratory tract infections including streptococcal infection with elevated ASO titers.[4] Endocapillary proliferation has been described in 20% and subepithelial humps in 41% of C3GN patients, both of which are common features of PIGN.
Etiologically, atypical PIGN and C3GN represent steps in a continuing spectrum. PIGN when triggered by an infection leads to activation of the AP which is quickly brought under control once infection abates.[3] In patients with a defect in AP regulation, there is continual AP activation with deposition of complement proteins and their breakdown products in the glomeruli, even after resolution of the infection; this leads to the development of “atypical” PIGN. If the defect is mild, AP control eventually occurs; resolving the glomerulonephritis. However, if the defect in AP regulation is severe in the form of mutations in complement-regulating proteins or autoantibodies to the C3 convertase; hematuria and proteinuria persist. Such a pathological pathway is similar to that seen in C3GN and represents the close conformity between these two diseases.
On ultrastructural examination, we identified TRIs in the endothelial cells. TRIs are most often seen in patients with lupus nephritis, HIV-associated nephropathy, and other viral infections. They are 20 to 28 nm subcellular structures with anastomosing membranous tubules in the cisternae of the endoplasmic reticulum; most commonly found in the cytoplasm of endothelial and lymphoreticular cells.[6] Their formation occurs in response to endogenous elevation of α and β interferons (IFNs) secondary to viral infections.[6] Evaluation of this patient did not reveal any evidence of viral disorders such as HIV, cytomegalovirus (CMV), adenovirus, or hepatitis viruses. Autoimmune serology was negative. The presence of TRIs in the absence of SLE (systemic lupus erythematosus) and HIV is rare and, to the best of our knowledge, has not been reported so far in the C3GN. In our case, they could possibly be a secondary marker of infection that may have possibly triggered overactivity of the AP.
In conclusion, our case represents the overlap of ultrastructural features seen in C3GN and DDD and raises the question of whether these features should be used to differentiate between C3GN and DDD. The presence of TRIs was a unique feature of our case.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Kidney disease caused by dysregulation of the complement alternative pathway: An etiologic approach. J Am Soc Nephrol. 2015;26:2917-29.
- [Google Scholar]
- Atypical postinfectious glomerulonephritis is associated with abnormalities in the alternative pathway of complement. Kidney Int. 2013;83:293-9.
- [Google Scholar]
- C3 glomerulonephritis: clinicopathological findings, complement abnormalities, glomerular proteomic profile, treatment, and follow-up. Kidney Int. 2012;82:465-473.
- [Google Scholar]
- Overlap of ultrastructural findings in C3 glomerulonephritis and dense deposit disease. Kidney Int. 2015;88:1449-50.
- [Google Scholar]
- Significance of tubuloreticular inclusions in the pathobiology of human diseases. Pathobiol Annu. 1976;6:221-57.
- [Google Scholar]




