

Translate this page into:
Heavy and Light chain amyloidosois presenting as complete heart block: A rare presentation of a rare disease
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Amyloidosis is an uncommon disease characterized by deposition of proteinaceous material in the extracellular matrix, which results from abnormal protein folding. Even though more than 25 precursor proteins are identified, majority of systemic amyloidosis results from deposition of abnormal immunoglobulin (Ig) light chains. In heavy chain amyloidosis (AH), deposits are derived from both heavy chain alone, whereas in heavy and light chain amyloidosis (AHL), the deposits are derived from Ig heavy chains and light chains. Both AH and AHL are extremely rare diseases. Here, we report an unusual presentation of IgG (lambda) AHL amyloidosis in the background of multiple myeloma, where the initial clinical presentation was complete heart block, which preceded the definitive diagnosis by 18 months.
Keywords
Complete heart block
heavy and light chain amyloidosis
multiple myeloma
systemic amyloidosis

Introduction
Heavy and light chain amyloidosis (AHL) is a rare disease. So far, only four case reports and one case series has been published.[1] Due to the paucity of data, not much is known about the biological and clinical behavior of AHL. Both AHL and AH are considered to have a better prognosis and considerably less cardiac involvement when compared with its more frequent counterpart, AL.
Case Report
A 57-year-old male presented with sudden onset of syncope and hypotension. His electrocardiogram showed third-degree heart block. He had normal serum creatinine, blood sugars and lipid profile. Urine examination showed trace proteinuria by dipstick. A 12-lead electrocardiogram revealed complete heart block with a ventricular rate of 40 beats/minute. Echocardiogram showed left ventricular hypertrophy with interventricular septal thickness of 26 mm; ejection fraction 55%, normal aortic, mitral, tricuspid and pulmonary valves, mild mitral regurgitation, and normal right ventricular pressures. Based on these findings, patient was diagnosed to have hypertrophic obstructive cardiomyopathy (HOCM) with complete heart block. The coronary angiogram showed normal coronaries. The patient received a dual chamber pacemaker following which he remained relatively asymptomatic; his systolic blood pressures documented during follow-up visits were consistently around 100 mm Hg. Nine months later, he noticed mild pedal edema and progressive numbness of toes, for which he did not seek medical attention.
He presented 18 months after the pacemaker implantation with worsening pedal edema shortness of breath and frequent episodes of syncope. On examination, he was found to have postural hypotension and bilateral symmetric peripheral neuropathy. The serum Cr was 2.2 mg/dl, serum protein levels were 7.4 g/dl with an albumin: globulin ratio of 2.9. Hemoglobin was 10 g/dl, peripheral smear showed increased rouleaux formation. His 24 h urine protein excretion was 2208 mg/day, urinary sediment was inactive. Ultrasound abdomen showed normal-sized kidneys. Nerve conduction study showed bilateral symmetrical sensory motor polyneuropathy. A 12-lead electrocardiogram showed that the pacemaker initiated rhythm in the ventricular demand mode with a rate of 60 beats/minute with intermittent under-sensing and a QRS amplitude of 5 mm in the limb leads. Serum electrophoresis revealed a monoclonal band in the region of gamma globulins and elevated free light chains (free kappa 39.4 mg/l, free lambda 136 mg/l) with a kappa/lambda ratio of 0.29. Bone marrow aspirate showed more than 30% plasma cells. A renal biopsy was performed.
Light microscopy (LM) revealed 27 glomeruli, of which 15 glomeruli were completely sclerosed. Viable glomeruli were enlarged in size and showed marked mesangial widening with prominent nodules composed of glassy, homogenous eosinophilic material, which was periodic acid-Schiff (PAS)-positive and silver-negative [Figure 1a and 1b]; many blood vessels showed similar material causing luminal occlusion. A prominent basement membrane thickening was observed. Interstitium showed moderate inflammatory infiltrate composed of lymphocytes and few plasma cells, and there was tubular atrophy amounting to 60% of the core. Immunofluorescence (IF) microscopy showed strong positive staining for heavy chain immunoglobulin (Ig) G and light chain lambda in the mesangium, along the glomerular basement membrane, tubular basement membrane as well as along the vessels [Figure 1c and 1d]. IgA, IgM, C3C, C1q and kappa were negative. The glomerular deposits revealed apple green birefringence on polarized microscopy [Figure 2a and 2b]. Electron microscopy (EM) revealed normocellular glomerulus with marked mesangial widening and numerous haphazardly arranged, nonbranching fibrils ranging from 7 nm to 10 nm in diameter [Figure 2c]. A repeat echocardiogram revealed concentric left ventricular hypertrophy, mild mitral regurgitation, mild tricuspid regurgitation, right ventricular systolic pressure of 44 mm Hg and a restrictive filling pattern [Figure 2d].
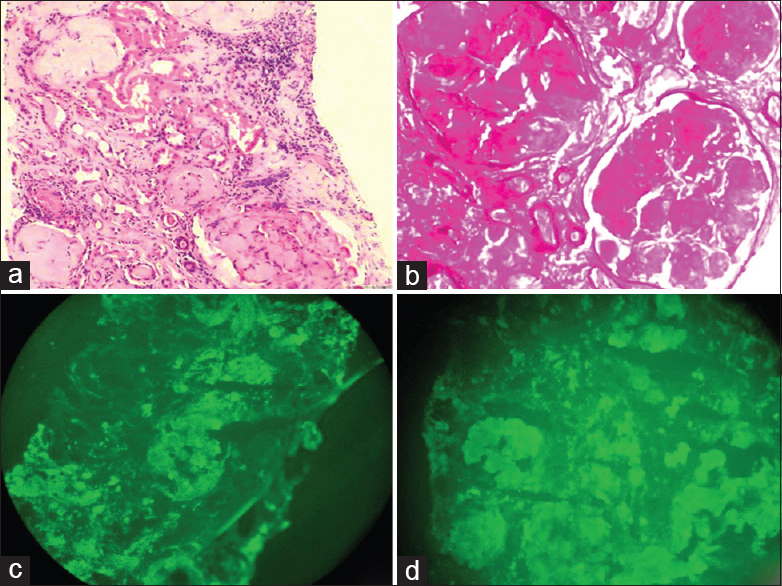
- (a) Glomeruli with marked mesangial widening, prominent nodules composed of glassy, homogenous, and eosinophilic material. In addition, one small arteriole shows luminal occlusion by similar material (H and E, ×200). (b) Glomeruli show periodic acid - Schiff (PAS) positive mesangial widening and nodules (PAS, ×400). (c and d) Immunofluorescence (IF) shows strong positivity for both IgG and lambda in the mesangiun, glomerular and tubular basement membrane (IF, ×400)
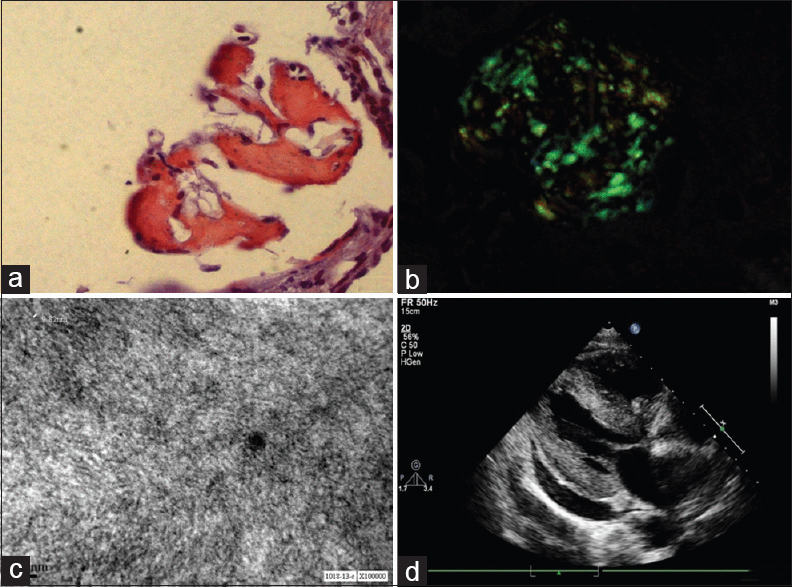
- (a and b) Congo red staining with apple green birefringence on polarized microscopy. (c) Characteristic amyloid fibrils ranging from 8 nm to 10 nm in diameter. (d) Echocardiogram showing left ventricular hypertrophy
Based on the above findings, the patient was diagnosed to have systemic amyloidosis (heavy and light chain) with multiple myeloma. Patient was started on bortezomib-based chemotherapy. Patient developed bronchopneumonia 2 weeks later and succumbed.
Discussion
Amyloidosis is characterized by extracellular deposition of insoluble 7-10 nm fibrils, having a beta-pleated ultrastructure. Systemic amyloidosis affects vital organs such as liver, kidney and heart. Ig-derived amyloid deposits accounts for about 85% of cases of systemic amyloidosis.[2] Of this majority is AL, characterized by Ig light chain deposition. On the other hand, AH and AHL are extremely rare. Both AL and AH/AHL can occur in association with plasma cell dyscrasias. There have been case reports of AHL occurring in the setting of lymphoma as well.[3]
There is only limited literature on the clinical characteristics and outcome of AHL. All reported cases of AHL except one had renal involvement.[3] Apart from case reports, there is only one systematic analysis of AH/AHL, which has compared the characteristics of this rare disease with the more common variant, AL.[1] Even though AHL has a similar age of presentation, gender predisposition and vital organ involvement, certain key differences from AL has been observed. When compared to AL, AHL has strikingly less cardiac involvement and comparatively better patient survival. Nasr et al. reported three patients with AH/AHL to have concurrent cardiac and renal involvement, of which two had AH whereas one had AHL.[1] There are no previous reports of AHL presenting with cardiac involvement with subsequent involvement of kidney. AHL is reported to have a considerably lesser diagnostic yield from abdominal fat pad and bone marrow biopsies. When compared to AL, majority of the patients will have an intact circulating monoclonal Ig as there is concomitant production of light as well as heavy chain. The monoclonal light and heavy chains are considered to be secreted by a single clone of B cells. A higher proportion of patients with AHL tend to have plasma cell dyscrasias when compared to AL.
Certain unique challenges can stand in the way of making a conclusive histologic diagnosis of AHL. The hallmark of renal amyloidosis is the presence of homogenous, amorphous, faintly eosinophilic PAS and silver-negative extracellular deposits on LM. This deposit exhibits a characteristic apple green birefringence on Congo red staining when viewed under polarized microscope. The deposits are composed of 7-10 nm fibrils, which can be visualized under EM. EM will reveal the fibrillary nature of the deposits, but is not useful for subtyping the amyloid. Atypical LM features have been described in around 25% of cases with AHL. These include PAS and silver-positive deposits, prominent glomerular basement membrane involvement and mesangial hypercellularity. These LM features are likely to be mistaken for fibrillary glomerulonephritis. In resource-poor settings where EM is not routinely employed, it is prudent to screen for Congo red positivity in all suspected cases of fibrillary glomerulonephritis, especially when it is associated with restriction of a heavy chain and/or light chain.[1] Renal biopsy of our patient also showed a significant amount of PAS positivity.
Most of the centers rely on IF microscopy for subtyping of renal amyloidosis. In AHL, IF microscopy will demonstrate restriction of a single Ig light chain and heavy chain, but can be negative in approximately 18% of cases.[2] The antibodies used in IF bind to the epitopes on constant domains of light and heavy chains. If these epitopes are deleted or significantly altered, the antibodies will not be able to bind. It is likely that a proportion of AHL amyloidosis may be missed if IF alone is used for amyloid typing. The demonstration of light and heavy chains may require additional testing by use of laser microdissection with tandem mass spectrometry (LMD/MS)-based proteomic analysis.[4] Employing LMD/MS as a tool for renal amyloid typing is likely to increase the diagnostic yield.[2] Another potential problem that may seriously affect the validity of IF is the nonspecific Ig and complement staining by amyloid deposits. This is encountered more frequently in AA amyloidosis than AL; this results from an in vitro phenomenon due to increased affinity to amyloid AA protein by the antibodies used or because of an in vivo stickiness of the amyloid to Igs.[5] If IF methods alone are used, the diagnosis of AHL should be limited to cases that show intense equal staining for a single heavy chain and one light chain.[1]
Cardiac involvement is common in AL but considered to be less common in AH/AHL. The classic clinical finding of low-voltage QRS complexes (limb leads <5 mm in height) with poor R wave progression in the anterior chest leads (pseudo infarction pattern) is described in 50% of patients in cardiac amyloidosis. Complete heart block is an uncommon presentation of amyloidosis; it has been described in <3% of patients who have cardiac amyloidosis. Typical echocardiographic findings include concentric ventricular thickening with right ventricular involvement, poor biventricular long-axis function with normal or near-normal ejection fractions. A speckled or granular myocardial appearance is considered to be characteristic, but seldom demonstrated and may depend on the machine gain settings.[6] The classic restrictive pattern, biventricular thickening and valvular infiltration occurs late in the course of amyloidosis. In 5% patients, an echocardiographic appearance indistinguishable from HOCM has been described.[7] The definitive diagnosis of cardiac amyloidosis is endomyocardial biopsy. Cardiac magnetic resonance imaging with gadolinium contrast is a useful noninvasive test, which could differentiate amyloidosis from other causes of left ventricular hypertrophy.[6]
Conclusions
To the best of our knowledge, this is the first report of AHL from India. We wanted to highlight the atypical presentation of complete heart block, which is a rare initial manifestation of AHL. The initial echocardiographic appearance was indistinguishable from HOCM; a restrictive pattern classical of amyloidosis was demonstrated fairly late in the course of the disease. Amyloidosis, even though uncommon, needs to be considered strongly in elderly patients presenting with left ventricular hypertrophy in the absence of hypertension. Another initial diagnostic dilemma in this patient was the atypical findings on LM. The renal biopsy showed significant PAS positivity, a finding, which is unusual in amyloidosis. Considering the fact that a major proportion of AH/AHL exhibit atypical features on LM, Congo red screening should be considered for amyloid-like deposits even if it is PAS/silver-positive. In view of the intrinsic limitations of IF in subtyping amyloid, it is possible that AHL might be underdiagnosed. Incorporating a direct protein identification method like LMD/MS is likely to improve the diagnostic yield.
Source of Support: Nil
Conflict of Interest: None declared.
References
- The diagnosis and characteristics of renal heavy-chain and heavy/light-chain amyloidosis and their comparison with renal light-chain amyloidosis. Kidney Int. 2013;83:463-70.
- [Google Scholar]
- Renal amyloidosis: Origin and clinicopathologic correlations of 474 recent cases. Clin J Am Soc Nephrol. 2013;8:1515-23.
- [Google Scholar]
- Co-deposition of amyloidogenic immunoglobulin light and heavy chains in localized pulmonary amyloidosis. Virchows Arch. 2005;447:756-61.
- [Google Scholar]
- Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood. 2009;114:4957-9.
- [Google Scholar]
- Typing of amyloidosis in renal biopsies: Diagnostic pitfalls. Arch Pathol Lab Med. 2007;131:917-22.
- [Google Scholar]
- Contemporary reviews in cardiovascular medicine - Management of the cardiac amyloidoses. Circulation. 2005;112:2047-60.
- [Google Scholar]




