

Translate this page into:
A Case of Posttransplant Fibrillary Glomerulonephritis
Address for correspondence: Dr. Shiva Kumar Ammayappan, Department of Nephrology, Super Speciality Block, PMSSY Building, Government Rajaji Hospital, Madurai Medical College, Madurai - 625 020, Tamil Nadu, India. E-mail: shiv.dr.0508@gmail.com
-
Received: ,
Accepted: ,
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Fibrillary glomerulonephritis (FGN) is a rare form of glomerulonephritis, usually occurring in concurrence with other conditions such as hepatitis C, dysproteinemia, autoimmune conditions, diabetes mellitus, and malignancy. The diagnosis is made by the presence of randomly oriented fibrillar deposits with a mean diameter of 20 nm, which stain positive for IgG and C3 and are negative for congo red and thioflavin T stains. Staining for DNAJB9 (DnaJ homolog subfamily B member 9) is a recently discovered mode of diagnosis of FGN without electron microscopy. The prognosis is poor and optimal treatment is yet not clearly defined, though rituximab may be useful in FGN patients with relatively preserved renal functions. In this case report, we discuss a case of post–renal transplant patient with de novo occurrence of fibrillary glomerulonephritis.
Keywords
Fibrillary GN
posttransplant
glomerulonephritis

Introduction
Fibrillary glomerulonephritis (FGN) is a rare proliferative form of glomerular disease. Pathologically, it is characterized by glomerular accumulation of randomly arranged fibrils with a diameter of about 20 nm. The striking pathologic resemblance to other forms of glomerulonephritis (GN) necessitates the use of DNAJB9 (DnaJ homolog subfamily B member 9) stain for the early and easier diagnosis of FGN. The occurrence of FGN in native kidneys is attributed to other concurrent conditions. In contrast, the occurrence of de novo FGN is very rare, and very few case reports have been published. Here, we discuss one such case.
Case Report
A 30-year-old male, who underwent a living-related donor kidney transplantation 7 years before, was admitted with graft dysfunction. His native kidney disease was not known. He was diagnosed at 16 years of age, when he was evaluated for anasarca. Renal biopsy was done, but the reports were not available and thus the native kidney disease is not known. He was treated conservatively for 6 years, after which he was initiated on hemodialysis. After 1 year of hemodialysis, he underwent a kidney transplant. He had attained immediate graft function, and his postoperative period was uneventful.
During his routine follow-up, he was found to have deranged renal function tests and nephrotic range proteinuria. He was on triple immunosuppression (tacrolimus, azathioprine, and prednisolone) and his tacrolimus trough levels were within normal limits. His creatinine was 2.7 mg/dL. Urine analysis showed loaded proteinuria and urine spot protein/creatinine ratio of 8. His complements were within normal limits and antinuclear antibody was negative. Serology for human immunodeficiency virus, hepatitis B, and hepatitis C were negative.
A transplant kidney biopsy was done for this patient. Light microscopy [Figure 1] showed global sclerosis in >50% of the glomeruli. Mesangial hypercellularity with matrix expansion was seen in all the viable glomeruli. Segmental endocapillary hypercellularity was seen in two and segmental sclerosis was seen in four glomeruli. Immunofluorescence showed a smudged positivity on the capillary loops and mesangium for IgG (+3) and C3 (+2) [Figure 2]. No light chain restriction was seen. Immunohistochemical stain for DNAJB9 was positive [Figure 2]. Thus, a diagnosis of the mesangial proliferative pattern of FGN was made. The patient was treated with rituximab, and the renal functions have been stable since then.
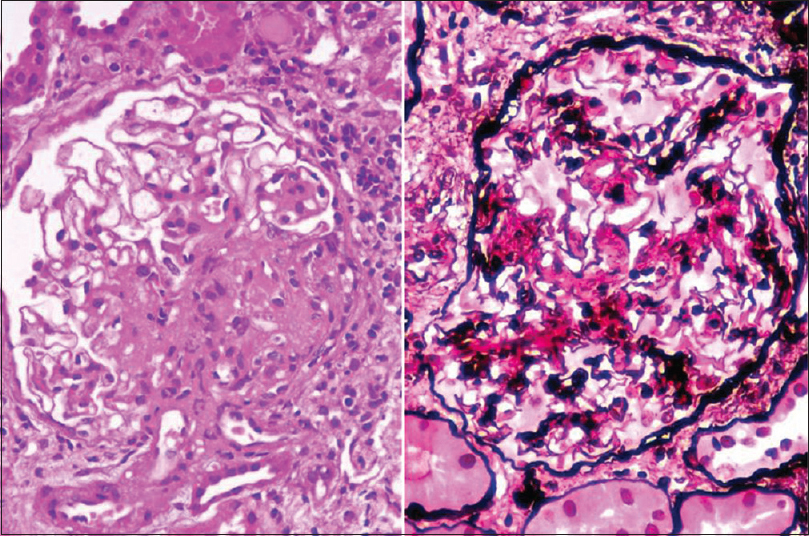
- Hematoxylin and eosin stain and PASM (periodic Schiff methenamine) stain

- Immunofluorescence for IgG and DNAJB9 stain
Discussion
FGN is a rare form of glomerular disease. The incidence of FGN is less than 1%.[1]
The mean age of presentation in native kidneys is in the sixth decade and is predominantly seen in females (66%). Very few cases of posttransplant FGN have been documented. There has been a report of de novo FGN due to chronic hepatitis C, where the primary renal disease was Type 1 membranoproliferative glomerulonephritis (MPGN).[2] In a multicentric study by Andeen et al.,[3] there were two cases of de novo FGN at 6 and 29 years posttransplant. Interestingly, both these patients had a renal failure due to polycystic kidney disease. Our patient is a male, is in his third decade of life, had a transplant 7 years ago with immediate graft function, and native kidney disease is not known.
Patients with FGN may present with renal insufficiency (70%), hematuria (82%), and nephrotic syndrome (36%).[4] Our patient had recent graft dysfunction (serum creatinine 2.9 g%) and nephrotic range proteinuria. Most cases are idiopathic, although association with other diseases is documented. The prevalence of hepatitis C virus infection in 7 to 27%, concurrent dysproteinemia in 13%, autoimmune diseases in 11%, diabetes mellitus in 24%, and malignancy in 9% of the patients with FGN has been described.[4] Our patient was screened for all the above-mentioned conditions, and thus was labeled idiopathic FGN.
There are five patterns of FGN on light microscopy.[1] The commonest pattern in most of the case series[5-7] is mesangial proliferative variant. The diffuse sclerosing pattern, which is characterized by >70% global glomerulosclerosis is associated with the worst prognosis. The rarest pattern is a membranous pattern. The mesangial proliferative variant and the membranous variant are associated with lower serum creatinine, less incidence of nephrotic syndrome, and long-term best prognosis. The variants associated with an intermediate outcome are membranoproliferative and diffuse proliferative patterns.
The immunofluorescence findings in FGN include ill-defined, “smudged” deposits that stain most intensely for IgG, usually accompanied by C3, kappa, and lambda. The deposits in FGN are nearly always present in a mesangial distribution.[4] IgG4 is the most common subtype of IgG present in the FGN deposits,[1] though IgG subtyping was not done in our patient. Studies done at the University of Washington[3] and Mayo clinic[8] identified DNAJB9 as one of the most abundant proteins in the FGN glomerular proteome, which was absent in glomeruli with amyloidosis, other forms of glomerulonephritis, or controls. They suggested that DNAJB9 may act as an autoantigen in FGN. The high specificity of the DNAJB9 immunohistochemical marker allows us to differentiate FGN from the above differentials in a more rapid and accurate way. Although electron microscopy (EM) was not done, the positive staining of DNAJB9 helped us in clinching the diagnosis of FGN.
In the normal kidney, DNAJB9 is present in renal tubular epithelial cells, podocytes, mesangial and endothelial cells, although at low levels. FGN is the only disease known to be associated with large amounts of extracellular deposition of DNAJB9.[4] Nasr et al.[7] established that staining for DNAJB9 was found to be 98% sensitive and >99% specific for the diagnosis of FGN. DNAJB9 staining was not observed in non-FGN glomerular diseases, except for very focal staining in one case of smoking-related glomerulopathy, who also had hepatitis C infection. It was also postulated that this case could represent a very early case of hepatitis C–associated FGN and as a result, the sample glomeruli for EM did not contain fibrillary deposition. The availability of immunohistochemical staining for DNAJB9 as a marker of FGN allows a rapid diagnosis and serves as a method for the diagnosis of FGN in many parts of the world, where EM is not available. DNAJB9 staining is now available worldwide. The stain used in the diagnosis of our patient was polyclonal IgG derived from rabbit and used 40% glycerol and phosphate-based buffer as a buffer. About 0.02% sodium azide is added as a preservative. Short-term storage can be done at +4°C but long-term storage is recommended at −20°C.
EM shows nonbranching, randomly oriented fibrils with a mean diameter of 20 nm (15–25 nm), which are nearly always present in the mesangial matrix, and in most cases, also permeate the lamina densa of the glomerular basement membrane, with minimal extension into the subendothelial or subepithelial regions. Extensive foot process effacement is seen in areas of fibril deposition. Granular electron-dense deposits are common in FGN, but typically are relatively focal in distribution.[4] In our patient, EM could not be done due to logistic reasons. Other differentials to be considered while making a diagnosis of FGN are immunotactoid glomerulonephritis (ITG) and amyloidosis. Amyloid fibrils are congo red positive and are smaller in diameter (about 10 nm).
Related to FGN but occurring even more rarely is ITG. In ITG, the fibrils are larger (>30 nm), hollow, and/or organized into bundles. Patients with ITG are more likely to have an underlying paraproteinemia or B-cell neoplasm, occurring in more than 50% of the cases.[2]
Rare cases of cryoglobulinemic GN form fibrils that have a similar ultrastructural appearance to FGN fibrils on EM.[4] Our patient was screened for serum cryoglobulins and rheumatoid factor, which were negative.
The definitive treatment of FGN is yet to be known. Immunosuppressives, in the form of steroids, cyclosporine, and cyclophosphamide, have been tried, albeit of no use. Hogan et al.[9] demonstrated the use of rituximab in FGN in 12 patients, the majority of whom had MPGN pattern on light microscopy. About 33% of the patients treated with rituximab showed a clinical response at a mean period of 40 months, particularly in patients with relatively normal baseline renal function. Our patient was treated with rituximab 2 months ago, and since then the renal functions have remained stable.
Five of the 14 patients who underwent a transplant in the Mayo case series had a disease recurrence at a mean posttransplant follow-up of 51 months.[6] Two patients in the Columbia series had no recurrence, at 4 and 8 years, although biopsy was not done.[1] Therefore, renal transplantation may offer a viable option in FGN, but the risk of recurrence is not negligible.
The prognosis of FGN is dismal. In the series from the Columbia University, the median time to reach end-stage renal disease was 24.2 ± 15.2 months, and the predictors of outcome were initial serum creatinine and the degree of interstitial fibrosis.[1] Our patient had renal failure in his second decade of life; therefore, the suspicion of native kidney FGN was low. Thus, we presumed our patient to have developed de novo allograft FGN of idiopathic origin.
In conclusion, FGN is a rare form of the glomerular disease characterized by randomly oriented, nonbranching fibrils with a mean diameter of 20 nm. The occurrence of de novo FGN is even rarer and needs to be differentiated from other forms of GN such as amyloid, cryoglobulinemic GN, and ITG. DNAJB9 is an excellent biomarker for FGN and has become the new gold standard for FGN diagnosis. Immunohistochemical staining of DNAJB9 has now replaced EM and allows for prompt diagnosis of FGN.[10] The prognosis remains poor, therapeutic options are still limited, and need to be addressed.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Fibrillary and immunotactoid glomerulonephritis: Distinct entities with different clinical and pathologic features. Kidney Int. 2003;63:1450-61.
- [Google Scholar]
- De novo fibrillary glomerulonephritis (FGN) in a renal transplant with chronic hepatitis C. Case Rep Transplant 2013 2013 978481
- [Google Scholar]
- DnaJ homolog subfamily B member 9 is a putative autoantigen in fibrillary GN. J Am Soc Nephrol. 2018;29:231-9.
- [Google Scholar]
- Morphologic and clinical features of fibrillary glomerulonephritis versus immunotactoid glomerulopathy. Am J Kidney Dis. 1993;22:367-77.
- [Google Scholar]
- Fibrillary glomerulonephritis: A report of 66 cases from a single institution. Clin J Am Soc Nephrol. 2011;6:775-84.
- [Google Scholar]
- DNAJB9 is a specific immunohistochemical marker for fibrillary glomerulonephritis. Kidney Int Rep. 2018;3:56-64.
- [Google Scholar]
- DnaJ heat shock protein family B member 9 is a novel biomarker for fibrillary GN. J Am Soc Nephrol. 2018;29:51-6.
- [Google Scholar]
- Rituximab treatment for fibrillary glomerulonephritis. Nephrol Dial Transplant. 2014;29:1925-31.
- [Google Scholar]
- Fibrillary glomerulonephritis and DnaJ homolog subfamily B member 9 (DNAJB9) Kidney 360. 2020;1:1002-13.
- [Google Scholar]




