

Translate this page into:
IgG4 related renal disease: A wolf in sheep's clothing
Address for correspondence: Dr. Rohan Augustine, No. 8, 1st C Main, Muthyalanagar, Mathikere P.O., Bengaluru - 560 054, Karnataka, India. E-mail: drrohan1996@rediffmail.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
IgG4 related disease is a fibro-inflammatory condition with involvement of renal and extra renal organs, characterized by lymphoplasmacytic infiltration with organ dysfunction. We describe three cases of IgG4 related renal disease from a tertiary care hospital in south India.
Keywords
IgG4 related renal disease
interstitial nephritis
storiform fibrosis

Introduction
IgG4-related renal disease is relatively a new entity attracting wide attention of the nephrology community. It is a fibro-inflammatory condition characterized by tumefactive lesions, dense lymphoplasmacytic infiltrate rich in IgG4-positive plasma cells, storiform interstitial fibrosis and elevated serum IgG4 concentration.[12] Autoimmune pancreatitis (AIP), the first entity under this group was described in 2001.[1] Since then, IgG4-related disease has been described in virtually every organ system: the biliary tree, salivary glands, periorbital tissues, kidneys, lungs, lymph nodes, meninges, aorta, breast, prostate, thyroid, pericardium and skin.[13456] IgG4-related kidney disease was first reported as an extra-pancreatic manifestation of AIP in 2004.[7] The renal presentation is primarily a tubulointerstitial nephritis although glomerular involvement has also been described. Recently, diagnostic criteria have been proposed by Kawano et al. [Table 1] based on the following conditions: (1) presence of kidney damage, (2) imaging showing enlarged kidneys with or without hypo or hyper echoic focal lesions, (3) serum IgG4 > 135 mg/dl, (4) characteristic renal histology showing storiform interstitial fibrosis with lympho-plasmacytic infiltration and (5) extra-renal pathology.[8]

Here, we describe three cases with different clinical manifestations and their diagnosis at our hospital, emphasizing that this disease exists even among our population.
Case Reports
Case 1
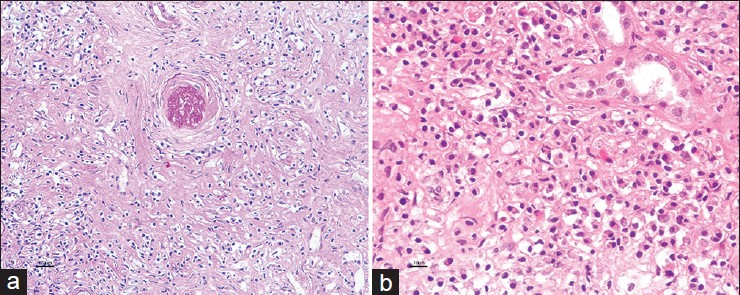
A 44-year-old male presented with dyspnea on exertion and cough for a few months. On radiological evaluation, he was found to have bilateral pneumonia with right sided pleural effusion. He was treated initially with broad-spectrum antibiotics and later empirically started on anti-tubercular drug therapy despite which his symptoms and radiological signs persisted. He was further investigated with a computed tomography (CT) scan of the chest, which revealed bilateral lung nodules with mediastinal lymphadenopathy. Ultrasonography of the abdomen revealed enlarged kidneys with tumefactive hyperechoiec areas [Figure 1] and CT abdomen revealed large kidneys with tumefactive hypodense lesions, hepatosplenomegaly with retroperitoneal and mesenteric lymphadenopathy. Fine needle aspiration cytology (FNAC) of mediastinal lymph nodes was reported as reactive hyperplasia. He then underwent laparoscopic biopsy of the liver and FNAC of the mesenteric nodes which revealed only reactive changes while the liver biopsy revealed dense chronic lymphoplasmacytic infiltrate with fibrosis and no granulomas to suggest tuberculosis (TB) or sarcoidosis. Blood chemistry revealed normal angiotensin converting enzyme levels, serum creatinine 2.2 mg/dl, total protein 9.2 g/dl, globulins 6.7 g/dl, hemoglobin 12.0 g/dl, erythrocyte sedimentation rate (ESR) of 56 mm in 1st hr. His serum electrophoresis showed significantly elevated gamma globulin fraction of 55.4% (range: 12-22%), but without an “M” spike. He underwent a kidney biopsy, which showed six glomeruli entrapped and surrounded by dense sclerotic interstitium with lymphoplasmacytoid infiltrates and numerous eosinophils [Figure 2]. The lymphoid cells were predominantly T-cells. The Bcl2 marker was negative and ki67 index was <5% thereby ruling out lymphoma. The immunoflouroscence study of the kidney biopsy was negative. The overall features were suggestive of inflammatory myofibroblastic tumour/pseudo lymphoma. After the renal biopsy, he was lost for follow-up for the next 2 years. He then got readmitted at our hospital with edema, shortness of breath. His serum creatinine was 7.7 mg/dl and he had signs of volume overload. He was dialyzed and a repeat renal biopsy was performed. The kidney biopsy revealed 16 glomeruli, which were seen entrapped and surrounded by a densely sclerotic interstitium, infiltrated by fibroblasts in a storiform pattern [Figure 3a]. The viable glomeruli were non-proliferative with patent capillary lumen. There were no necrotizing lesions or granulomas. The immunoflourescence study was negative. The immunohistochemistry (IHC) was positive for CD138 and revealed IgG4 positive plasma cells >40/hpf [Figure 3b]. His serum IgG4 level was 26.9 g/l (normal value <135 mg/dl) and the serum IgG4/IgG ratio was 68% (normal <5%). The diagnosis of IgG4 related real disease was established in him based on diagnostic criteria and he was started on oral prednisolone for the next 4 months. After 4 months of treatment with oral steroids, his serum creatinine decreased to 4.0 mg/dl and he became dialysis independent. He was continued on oral steroids in tapering doses with which he reports good improvement in his pulmonary symptoms and X-rays show resolution of his mediastinal nodes.

- Ultrasound showing focal hyperechoic tumefactive lesions in the kidneys with preserved kidney size
- (a) Bland interstitial fibrotic process displacing the tubules wide apart (Periodic acid–Schiff, ×20), (b) Inflammatory infiltrates of dense plasma cells, eosinophils and lymphocytes (H and E, ×40)

- (a) Expansile fibroinflammatory process with typical storiform pattern of fibrosis entrapping the obsolescent glomerulus and accompanied inflammatory cell components (MT, ×10), (b) Immunoperoxidase stain shows significant increase in IgG4+ cells, >40/hpf (×20)
Case 2
A 57-year-old male with long standing hypertension and a past history of transurethral resection of prostate for benign prostatic hyperplasia presented with azotemia. His serum creatinine at the time of his prostate surgery was 1.3 mg/dl and the histopathology of the prostate was suggestive of eosinophilic prostatitis. At presentation, he was found to have serum creatinine of 2.5 mg/dl with a bland urinary sediment. Ultrasonography of the abdomen revealed right kidney 11.9 cm, left kidney 11.8 cm in size with tumefactive hyperechoiec lesions in both kidneys. His hemogram, liver functions were normal except for increased serum globulins and his serum electrophoresis showed increased gamma globulin fraction of 47% (range: 12-22%) without a monoclonal “M” spike. He underwent a kidney biopsy, which on light microscopy showed normal glomerular morphology with a densely sclerotic interstitium infiltrated by fibroblasts in a storiform pattern and dense lymphoplasmacytic infiltrate with numerous eosinophils. The immunofluoroscence did not reveal any immune complex deposits in the glomeruli although there were IgG (2+), C1q (2+), kappa (2+) and lambda (2+) deposits along the tubular basement membranes. The imunohistochemistry revealed strong positivity for IgG4 plasma cells (>25/hpf). His total serum IgG levels were 1980 mg/dl (mormal 550-1900mg/dl) with serum IgG4 level being 10.4 g/l (normal <135 mg/dl). The serum IgG4/IgG ratio was 52% (normal <5%). The diagnosis of IgG4 related renal disease was confirmed and he was treated with oral prednisolone 0.6 mg/kg (40 mg once daily). His serum creatinine reached a nadir of 1.8 mg/dl after 3 weeks of treatment with oral steroids. Subsequently, he was given mycophenolate mofetil and steroids were stopped. After 3 months of treatment, his serum IgG levels were 967 mg/dl (550-1900mg/dl) and IgG4 was 1.22 g/l.
Case 3
A 62-year-old man with long standing diabetes and hypertension presented with symptoms of generalized weakness and progressive weight loss. His physical examination was unremarkable and he had no peripheral lymphadenopathy or organomegaly. His urine analysis was unremarkable. His blood chemistry revealed hemoglobin 8.6 g/dl, ESR 115 mm in 1st hr, creatinine 3.8 mg/dl, globulin 4.5 g/dl. His serum protein electrophoresis revealed increased gamma globulin fraction of 45%, but did not reveal an “M spike.” Ultrasonography revealed bulky kidneys with increased cortical echogenicity. There was no evidence of intra-abdominal lymphadenopathy or hepatosplenomegaly. Kidney biopsy revealed normal glomerular morphology with dense lymphoplasmacytic interstitial infiltrate with fibrosis, though there was no evidence of a classic storiform pattern. Immunofluoroscence revealed no immune deposits in the glomeruli and there were IgG (1+), C3 (1+), C1q (1+), kappa (1+) and lambda (1+) deposition along the tubular basement membranes. IHC revealed predominant IgG4 positive plasma cells (>35/hpf) and markers for malignancy were negative. His Serum IgG level was 3260 mg/dl (550-1900) and serum IgG4 level was 20.6 g/l (normal <135 mg/d). The serum IgG4: IgG ratio was 63% (normal <5%). This patient also met the criteria for the diagnosis of IgG4 related renal disease. He was started on steroids for the same, but was lost to follow-up.
Discussion
IgG4-related renal disease is a relatively new clinical entity. It is primarily a tubulointerstitial renal disease, which predominantly affects males. The key morphologic features are a dense lymphoplasmacytic infiltrate organized in a storiform (i.e., matted and irregularly whorled) pattern with mild-to-moderate eosinophilic infiltrates.[2910] The inflammatory lesion frequently forms a tumefactive mass that destroys the involved organ and granulomas are unusual. IHC staining for IGg4 plasma cells are essential to confirm the histological diagnosis. A variety of cut-off points, ranging from >10 to >50 IgG4-positive plasma cells/hpf along with a ratio of IgG4/IgG bearing plasma cells of >50% strongly suggest the diagnosis.[111213] The systemic manifestations such as organomegaly, mediastinal lymphadenopathy, radiological tumefactive lesions in the kidney, renal dysfunction with sub-nephrotic proteinuria and a bland sediment should alert the clinician to suspect IgG4 related renal disease. Early diagnosis and treatment with steroids provides the best chance for renal survival.
The pathophysiology of this disease is poorly understood. Many factors, namely genetic, bacterial infection with Helicobacter pylori, molecular mimicry and abnormal autoimmune response have been implicated.[141516] It is regarded that T-helper (Th2) cell mediated immune response predominates in IgG4-related disease.[17] Another immunologic characteristic is the activation of regulatory T (Treg) cells in contrast to other classic autoimmune conditions, in which the function of Treg cells is impaired.
The closest histopathological mimicks of IgG4-related disease are lymphomas. The clonality studies differentiate it from lymphoma. B-cell lymphomas show the presence of a predominantly B-cell infiltrate; in contrast to the primary T-cell lymphoid infiltrate in IgG4-related disease. Another mimic of IgG4 related diseases are other inflammatory lesions. Patients with IgG4-related disease show diffuse infiltrates of IgG4-bearing plasma cells in contrast to focal aggregates seen in inflammatory conditions.
There are no randomized controlled trials regarding treatment for IgG4 related kidney disease. Glucocorticoids are the initial therapy of choice. A consensus statement suggested treating patients with prednisolone at a dose of 0.6 mg/kg body weight/day for 2-4 weeks followed by tapering the dose to 5.0 mg/day over a period of 3-6 months and then continuing steroids at a dose between 2.5 and 5.0 mg/day for up to 3 years.[181920] Another approach has been to discontinue glucocorticoids at the end of 3 months and using steroid sparing immunosuppressive agents such as azathioprine, mycophenolate mofetil and methotrexate to maintain remission. In patients who relapse or are refractory to steroids and immunosuppressive drugs, B-cell depleting agents such as rituximab may be the alternative choice.[2122] The major predictor of treatment response is the degree of fibrosis.
The clinical presentations in these three cases were varied. The first patient presented with pneumonia and mediastinal lymphadenopathy. He was initially treated as bacterial pneumonia with antibiotics and later treated as TB. He was suspected to have either lymphoma or sarcoidosis since he had mediastinal and abdominal lymphadenopathy with organomegaly. A specific diagnosis could not be reached despite FNAC of the mediastinal nodes, biopsy of mesenteric lymph nodes and a liver biopsy. TThe liver biopsy did show a dense chronic inflammation with predominant lymphocytes and plasma cells effacing the liver architecture, which in hind-sight is consistent with IgG4 related disease, but was not suspected at that point in time. The ultrasonography and CT abdomen did show some typical features of IgG4 related renal disease, but was not suspected as we had no similar precedents in the past. Although, the first renal biopsy showed a densely sclerotic interstitium with lymphoplasmacytoid and eosinophilic infiltrates meeting the criteria for IgG4 related renal disease, it was not suspected as our awareness of this new entity was primitive. By the time the second renal biopsy was performed, the clinical, biochemical and histological features of the disease were well-defined enabling us to confidently make a diagnosis of IgG4 related renal disease. Awareness of this new disease entity also led us to quantify IgG and IgG4 and also performing IHC for IgG4 cells in the biopsy. At the time of presentation, he needed dialysis since he had severe renal failure and volume overload. He achieved a partial response to corticosteroids and became dialysis independent with significant improvement in his pulmonary symptoms and resolution of mediastinal lymphadenopathy. The second patient presented with renal azotemia with no constitutional symptoms. He neither had lymphadenopathy nor organomegaly. His kidneys were bulky with tumefactive lesions. The renal histology was typical of IgG4 related renal disease with high serum IgG4 levels. He responded well to oral prednisolone with improvement in renal function and reduction in IgG and IgG4 fractions. The third patient presented with constitutional symptoms and renal azotemia. He did not have lymphadenopathy or organomegaly. He had bulky and hyperechoiec kidneys with an unremarkable urine analysis. The renal biopsy showed typical histology of IgG4 related kidney disease with elevated IgG and IgG4 fractions. He was treated with oral corticosteroids, but was lost for follow-up. The first case primarily presented as an extra renal disease that later developed renal involvement. The second and third cases presented primarily as a renal disease. The diagnosis in the first case was delayed because our awareness of IgG4 related disease was poor. The diagnosis was fairly rapid in second and third cases as our index of suspicion became higher.
There have only been isolated case reports of IgG4 related renal disease from India in the past.[23] The present case series adds to the growing literature from our country regarding this clinical condition.
Summary
IgG4 related renal disease can manifest as part of a multisystem disease or primarily as a renal disease. Renal involvement presents predominantly as an interstitial nephritis. A multisystem disease with involvement of lung, liver, spleen, lymph nodes and renal dysfunction with minimal proteinuria and bland urine sediment should evoke the clinical suspicion of IgG4 related disease. The typical radiological findings of tumefactive lesions in the kidney should further alert the clinician to suspect this disease and offer a renal biopsy. The ultrasonographic findings in the kidney may mimic a lymphoma. Awareness of this clinical entity would enable the clinician to have a high index of suspicion. The typical storiform pattern of interstitial fibrosis with dense plasma cell infiltrate in the interstitium should alert the nephropathologist to perform IHC stain for IgG4 plasma cells. Subsequently, requesting for serum IgG4: IgG ratio will further help to reach a definitive diagnosis. Early diagnosis and therapy is vital for the recovery of renal functions and overall prognosis.
Source of Support: Nil
Conflict of Interest: None declared.
References
- A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol. 2003;38:982-4.
- [Google Scholar]
- IgG4-related systemic disease and lymphoplasmacytic aortitis. Arthritis Rheum. 2009;60:3139-45.
- [Google Scholar]
- Riedel's thyroiditis and multifocal fibrosclerosis are part of the IgG4-related systemic disease spectrum. Arthritis Care Res (Hoboken). 2010;62:1312-8.
- [Google Scholar]
- Lymphoplasmacytic infiltration of multiple organs with immunoreactivity for IgG4: IgG4-related systemic disease. Intern Med. 2006;45:163-7.
- [Google Scholar]
- Autoimmune pancreatitis and IgG4-related sclerosing disease. Nat Rev Gastroenterol Hepatol. 2010;7:401-9.
- [Google Scholar]
- High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344:732-8.
- [Google Scholar]
- Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011;15:615-26.
- [Google Scholar]
- Subclassification of autoimmune pancreatitis: A histologic classification with clinical significance. Am J Surg Pathol. 2011;35:26-35.
- [Google Scholar]
- IgG4-related disease: A cross-sectional study of 114 cases. Am J Surg Pathol. 2010;34:1812-9.
- [Google Scholar]
- Use of immunohistochemistry for IgG4 in the distinction of autoimmune pancreatitis from peritumoral pancreatitis. Hum Pathol. 2010;41:643-52.
- [Google Scholar]
- Autoimmune pancreatitis: A systemic immune complex mediated disease. Am J Surg Pathol. 2006;30:1537-45.
- [Google Scholar]
- Diagnosis of autoimmune pancreatitis using its five cardinal features: Introducing the Mayo Clinic's HISORt criteria. J Gastroenterology. 2007;42(Suppl 18):39-41.
- [Google Scholar]
- HLA DRB10405-DQB10401 haplotype is associated with autoimmune pancreatitis in the Japanese population. Gastroenterology. 2002;122:1264-9.
- [Google Scholar]
- Substitution of aspartic acid at position 57 of the DQbeta1 affects relapse of autoimmune pancreatitis. Gastroenterology. 2008;134:440-6.
- [Google Scholar]
- Helicobacter pylori and autoimmune pancreatitis: Role of carbonic anhydrase via molecular mimicry? J Cell Mol Med. 2005;9:741-4.
- [Google Scholar]
- Possible involvement of T helper type 2 responses to Toll-like receptor ligands in IgG4-related sclerosing disease. Gut. 2010;59:542-5.
- [Google Scholar]
- Research Committee for Intractable Pancreatic Disease and Japan Pancreas Society. Japanese consensus guidelines for management of autoimmune pancreatitis: III. Treatment and prognosis of AIP. J Gastroenterol. 2010;45:471-7.
- [Google Scholar]
- Immunoglobulin G4-associated cholangitis: Clinical profile and response to therapy. Gastroenterology. 2008;134:706-15.
- [Google Scholar]
- Rituximab for the treatment of IgG4-related disease: Lessons from 10 consecutive patients. Medicine (Baltimore). 2012;91:57-66.
- [Google Scholar]
- Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 2010;62:1755-62.
- [Google Scholar]
- IgG4 related interstitial nephritis: A case report and review of literature. Indian J Nephrol. 2013;23:308-11.
- [Google Scholar]




