

Translate this page into:
Proliferative glomerulonephritis with monoclonal immunoglobulin deposition disease: The utility of routine staining with immunoglobulin light chains
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Proliferative glomerulonephritis occurring as a consequence of monoclonal glomerular deposits of IgG is uncommon. It is a form of renal involvement in monoclonal gammopathy that mimics immune complex glomerulonephritis. Here, we report the first series of proliferative glomerulonephritis with monoclonal IgG deposits (PGNMID) from the Indian subcontinent highlighting use of light chain immunofluorescence (IF) in routine renal biopsy interpretation. We retrieved 6 patients diagnosed as proliferative glomerulonephritis with monoclonal IgG deposits (PGNMID) out of 160 biopsies (3.7%) with membranoproliferative patterns over 5 1/2 years (2009–2014), one of whom had recurrence 6 months post-renal transplant. Four (67%) patients presented with rapidly progressive renal failure and two (33%) with nephrotic syndrome. None of these patients had overt multiple myeloma. The predominant histologic pattern was membranoproliferative with all the biopsies showing IgG3 Kappa deposits on IF. The deposits were primarily subendothelial on electron microscopy.
Keywords
Kappa
lambda
monoclonal IgG
proliferative glomerulonephritis

Introduction
Renal involvement in monoclonal plasma cell disorders is heterogeneous with those manifesting as monoclonal glomerular deposits of IgG being relatively uncommon. Renal diseases caused by monoclonal IgG deposition include light- and heavy-chain deposition disease (LHCDD), type 1 cryoglobulinemic glomerulonephritis, immunotactoid glomerulonephritis (IT), light- and heavy-chain amyloidosis, and rarely fibrillary glomerulonephritis. In 2004, Nasr et al. reported a novel form of glomerular injury related to monoclonal IgG deposition, which they termed “proliferative glomerulonephritis with monoclonal IgG deposits” (PGNMID).[1] Subsequently several studies have described this entity, with the largest series by Nasr et al. themselves (37 cases).[234] A total of 68 cases of PGNMID have been reported till date of which 10 were diagnosed in renal allografts.[56] Here we report the first series of PGNMID in Indian subcontinent.
Materials and Methods
Six patients with PGNMID have been reported among all native and transplant renal biopsies received at PGIMER, Chandigarh, from 2009 through February 2014 (51/2 years). They comprised 3.7% of the biopsies having membranoproliferative pattern (n-160). The diagnostic criteria used for PGNMID[5] were renal biopsy finding of glomerulonephritis with presence of the following: (1) glomerular immune deposits staining positive for γ heavy chain (IgG), with negative stain for α (IgA) and μ (IgM) heavy chains, indicating restriction to a single (γ) Ig class; (2) positive staining for a single γ (IgG) subclass (IgG1, IgG2, IgG3, or IgG4); (3) positive staining for a single light-chain isotype (κ or λ), indicating monoclonality; (4) predominantly granular electron-dense deposits in mesangial, subendothelial, and/or subepithelial locations by electron microscopy (EM), resembling immune complex glomerulonephritis; and (5) no clinical or laboratory evidence of cryoglobulinema.
Renal biopsy samples were processed by standard techniques for light microscopy (LM), immunofluorescence (IF), and EM. IF was performed on 3-μm cryostat sections using polyclonal fluorescein (FITC)-conjugated antibodies to IgG, IgM, IgA, C3, C1q, and κ and λ light chains (Dako). Determination of the IgG subclass was performed using monoclonal FITC-conjugated antibodies to IgG1 (clone 8c/6-39), IgG2 (clone HP-6014), IgG3 (clone HP-6050), and IgG4 (clone HP-6025). IF staining intensity was graded 0-3+ on a semi-quantitative scale.
Patients' medical records were reviewed for demographic information, presenting clinical and laboratory findings, presence of serum and urine M-spike, treatment, and outcome. The following definitions were applied:
-
Renal insufficiency: Serum creatinine >1.2 mg/dl
-
Hematuria: >5 red blood cells per high-power field on microscopic examination of the urinary sediment
-
Nephrotic syndrome was defined as 24-h urine protein ≥3.5 g/d, hypoalbuminemia (serum albumin <3.5 g/dl), and peripheral edema
-
Rapidly progressive renal failure (RPRF): Rapid deterioration in renal function over a period of 2 weeks to 3 months
-
Complete remission (CR): Remission of protienuria to <500 mg/d with normal renal function
-
Partial remission (PR): Reduction in proteinuria by at least 50% and to <2 g/d with stable renal function (no more than a 20% increase in serum creatinine)
-
Persistent renal dysfunction (PRD): Failure to meet criteria for either CR or PR, but not reaching end stage renal disease (ESRD), including patients with unremitting proteinuria or progressive chronic kidney disease
-
End stage renal disease (ESRD): Requiring renal replacement therapy for >3 months.
Results
Clinical and laboratory features
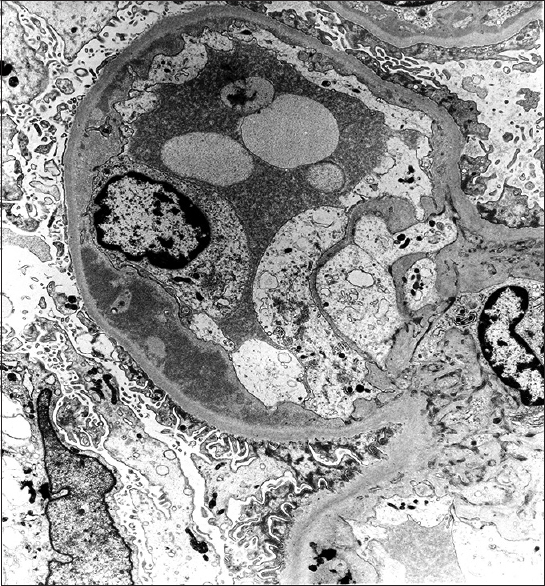
Five out of six patients (83%) were males [Table 1], with a mean age of 50 years (range 38–75 years). Four (67%) patients presented with RPRF and two (33%) with nephrotic syndrome. Peripheral edema was present in 4 (67%) patients. All the patients at presentation had proteinuria with a mean 24-h urine protein of 3.47 g (range 2.1–4.8 g). Proteinuria was in the nephrotic range in 4 (67%) patients. Micro-hematuria was documented in all patients, with none of them having gross hematuria. Hypertension was observed in four patients (67%). Four (67%) patients had renal insufficiency, including three (50%) patients requiring dialysis. The mean serum creatinine was 3.70 mg/dl (range 1.3–5.8 mg/dl). The mean serum albumin was 2.06 g/dl (range 1.6–2.4 g/dl).
None of the patients had monoclonal proteins detectable on standard serum protein electrophoresis and urine protein electrophoresis. Serum free light-chain (sFLC) assay was done in all the patients. Although, sFLC ratio was within the normal range (reference range for the sFLC ratio in patients with kidney failure is 0.37–3.1), all of them had kappa dominant light chain synthesis [Table 1]. Bone marrow examination performed revealed mean plasma cells of 2% (range 2–3%). None of the patients had lymphadenopathy, hepatosplenomegaly or lymphoma.
Serum cryoglobulin titers were negative, and none of the patients had any systemic manifestations of cryoglobulinema. There was no history of recent or chronic infection, systemic lupus erythematosus, rheumatoid arthritis, mixed connective tissue disease, or Sjögren syndrome in any patient.
Renal biopsy findings
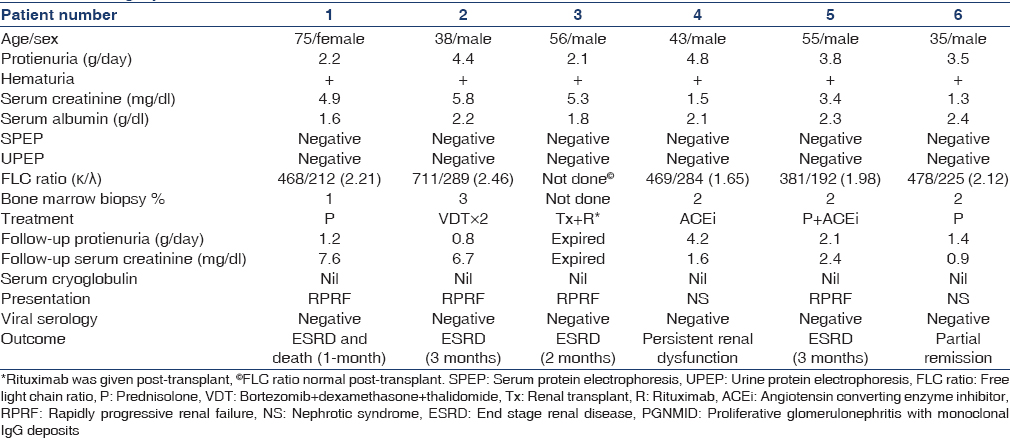
The kidney biopsies included mean of 15.5 glomeruli (range 8–42 glomeruli), of which on an average 4.2 glomeruli were globally sclerotic [Table 2]. The histologic pattern, seen was membranoproliferative glomerulonephritis in all cases (100%) characterized by diffuse and global double-contoured glomerular capillary walls with mesangial cell interposition and mesangial expansion by increased mesangial cell number and matrix [Figure 1]. Segmental endocapillary proliferation was present in four (67%) cases. None of the cases showed crescents. One (16%) patient had in addition features of diabetic glomerulosclerosis (Case 3). The degree of tubular atrophy and interstitial fibrosis was moderate and severe in 2 cases each while it was mild and absent in 1 case each. Arteriosclerosis ranged from mild (n-2) to moderate (n-2) to severe (n-2). Interstitial inflammation ranged from mild (n-2) to moderate (n-4).

- Photomicrograph (case 3) showing MPGN pattern with segmental endocapillary proliferation (PAS, ×40). The glomerulus showed deposits of IgG and kappa only along glomerular capillary loops and in mesangium (direct immunofluorescence, fluorescein, IgG, kappa, lambda, ×40)
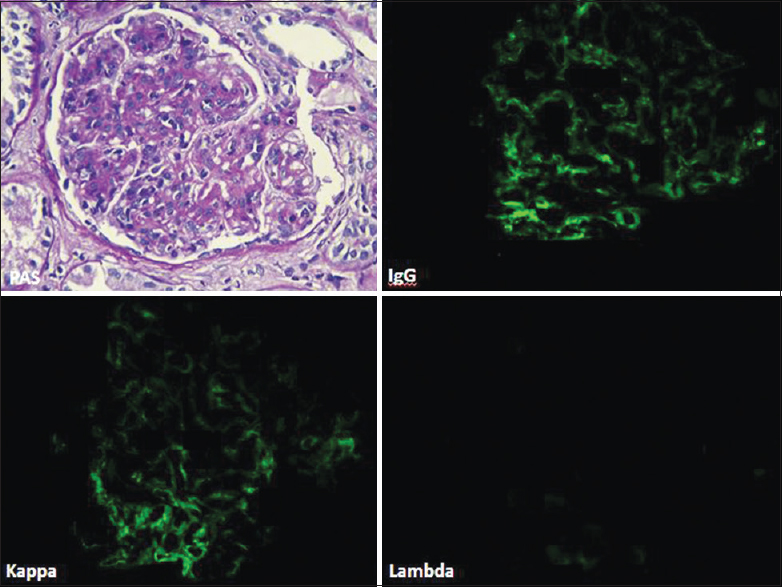
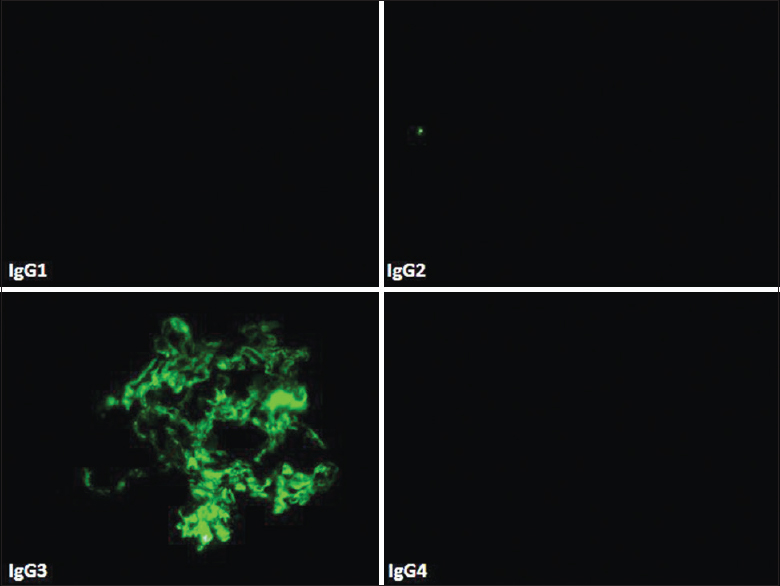
Glomeruli showed granular staining for IgG and C3 along glomerular capillary loops and focally in mesangium with light chain restriction (kappa restricted in 6 cases) by direct IF. IF for IgG subtypes demonstrated presence of IgG3 alone in all the 6 cases [Figure 2]. EM confirmed presence of subendothelial deposits with effacement of foot process [Figure 3]. Some deposits in mesangial regions were seen in all cases [Figure 3]. Absence of Randall type of deposits excluded monoclonal immunoglobulin deposition disease (light/heavy chain) and absence of organized deposits excluded cryoglobulinemic glomerulonephritis and immunotactoid glomerulonephritis.
- Direct immunofluorescence (case 3) showing IgG subtyping with IgG3 deposits only with absence of other three subclasses of IgG (fluorescein, IgG1, IgG2, IgG 3, IgG4, ×40)
- Electron microscopy (case 3) showed subendothelial electron dense deposits with effacement of foot processes of podocytes (Uranyl acetate, ×17,100)
Treatment
Prednisolone alone was given to two (32%) patients, one of whom had PR and other progressed to ESRD. Prednisolone with angiotensin converting enzyme inhibitor (ACEi) was administered to one (16%), who progressed to ESRD in 3 months. One (16%) patient was given Bortezomib, Dexamethasone, Thalidomide (VTD) regimen and developed ESRD in 3 months. One of the cases (Case 3) was diagnosed in the native kidney in retrospect, following the detection of the disease in the allograft biopsy. He was given Rituximab post-renal transplant and died of pulmonary infections 6 months later. One patient (Case 4) has received ACEi only without any immunosuppressive therapy despite advice and continues to have PRD.
Clinical outcome
Clinical follow-up was available for 6 patients [Table 1]. On follow-up, one (16%) patient (case 6) had PR, one (16%) patient (case 4) had PRD, and four patients (67%) had progressed to ESRD over a period of 3 months. Two (33%) patients died, one of whom died 6 months post-renal transplantation.
Discussion
The immune complex-mediated glomerulonephritis are characterized by localization in the glomeruli of antibodies complexed to endogenous or exogenous antigens, which at the ultrastructural level appear as granular electron-dense deposits. Morphologic patterns could be endocapillary proliferative glomerulonephritis, membranoproliferative glomerulonephritis or mesangioproliferative glomerulonephritis. The immunoglobulins deposited are typically polyclonal. In cases of endocapillary proliferative or membranoproliferative glomerulonephritis in which the deposits stain for IgG and a single light chain, diagnostic considerations would include PGNMID, type 1 cryoglobulinemic glomerulonephritis, LHCDD and immunotactoid glomerulonephritis. This distinction can only be made by routinely using immunoglobulin light chains in IF panel. Otherwise, all these cases will be diagnosed as immune complex-mediated MPGN (MPGN type 1). Once the diagnosis of monoclonal glomerular lesion is established, these can be further distinguished by electron microscopic examination by type of deposits, that is, granular electron-dense immune complex type of deposits in PGNMID (similar to MPGN Type 1), Randall type of deposits in monoclonal immunoglobulin deposition disease (light/heavy chain) and organized deposits in cryoglobulinemic glomerulonephritis and immunotactoid glomerulonephritis. Based on the diagnostic criteria for PGNMID[1] proposed by Nasr et al. the other diagnoses should be carefully excluded. All six cases in the present series fulfilled the criteria and were hence categorized under PGNMID.
Proliferative glomerulonephritis with monoclonal IgG deposits has been documented in adults with a mean age of 55 years[1] and more often in females. Although the mean age in the present cohort was also 50 years; however, majority (83%) of present patients were males, which probably is a matter of chance. Most patients of PGNMID present with nephrotic-range proteinuria, hematuria with or without renal insufficiency, whereas only two (33.3%) patients in present cohort had nephrotic syndrome, while hematuria and renal insufficiency were seen in all 6 of them. Of the 68 cases of PGNMID reported till date, only 4.4% were associated with hematologic malignancy (1-multiple myeloma, 2-chronic lymphocytic leukemia).[14] None of the cases in the present series had associated hematologic malignancy. However, sFLC assay done in six patients showed increased serum kappa levels in all of them (reference FLC ranges in renal failure: κ−3 = 251 mg/L and λ−1 = 251 mg/L), even though the sFLC ratio was within normal range in all of them.
The prognosis of this disease in the native kidney is variable. On the follow-up available in 32 of 37 previously reported patients, 37.5% recovered renal function, 37.5% developed PRD, and 22% progressed to ESRD.[1] In the current study, four patients progressed to ESRD in a duration of 3 months, 1 (16%) of whom received renal transplant (Case 3) and had recurrence of the disease. He received Rituximab therapy and died due to pneumonia. One patient has shown PR and is currently on follow-up. Interestingly one patient (Case 4) has received only ACE inhibitors and has refused any other therapy despite advice. While he continues to have PRD, no progression has been noted over a period of 2 years. In comparison to 22% patients progressing to ESRD in the report by Nasr et al. 4 (67%) patients in the current series progressed to ESRD. Although the number of cases is less to draw any conclusions, higher rates of progression in this series might be due to late presentation and poor compliance with therapy as a result of financial constraints of the patients.
One of the cases (Case 3) was diagnosed in the native kidney in retrospect, following the detection of the disease in the allograft biopsy. The patient was a diabetic and linear accentuation of lambda light chains noted on IF of native kidney was mistaken for granular staining, thus missing the monoclonality. This case emphasizes the need for routine use of kappa and lambda in IF for detection of cases of PGNMID, which otherwise would be missed, especially in India where routine use of light chains is still not a common practice in many centers, Nine cases of recurrent PGNMID have been described after kidney transplantation.[5678] These cases seem to have a better prognosis compared to the disease in native kidneys, probably due to the use of immunosuppressive therapy in transplant recipients.[4]
Although the exact pathogenesis of PGNMID remains elusive, it is proposed to be due to clonal proliferation of B-lymphocytes or plasma cells that hypersecrete abnormal IgG capable of self-aggregation and deposition in the glomerulus as electron-dense deposits. It is possible that the abnormal clone arises secondary to normal immune responses.[1] IgG3, the most common IgG subtype in PGNMID, is thought to be particularly nephritogenic with ability to self-aggregate and fix complement, resulting in the influx of inflammatory cells and subsequent proliferative GN.[2910] Not surprisingly all 6 of our cases had IgG3 deposits.
Conclusions
Proliferative glomerulonephritis with monoclonal IgG deposits cases in the current series are clinically and pathologically similar to the ones reported in the literature except for very poor renal outcome in most of the cases. Because the majority of patients do not have M-spike or plasma cell dyscrasias, renal biopsy with careful attention to light chain and IgG isotype staining is essential for diagnosis. Electron microscopic evaluation is mandatory to differentiate between the monoclonal glomerular proliferations. Larger multicenter studies of PGNMID will be required to define the exact nature of PGNMID in India and devise optimal therapeutic approach. In the case of renal allograft, use of protocol biopsies would allow us to make an earlier histologic diagnosis of recurrent PGNMID.
Source of Support: Nil
Conflict of Interest: None declared.
References
- Proliferative glomerulonephritis with monoclonal IgG deposits: A distinct entity mimicking immune-complex glomerulonephritis. Kidney Int. 2004;65:85-96.
- [Google Scholar]
- Proliferative glomerulonephritis with monoclonal IgG deposits. J Am Soc Nephrol. 2009;20:2055-64.
- [Google Scholar]
- Characteristics of proliferative glomerulo-nephritis with monoclonal IgG deposits associated with membranoproliferative features. Clin Nephrol. 2009;72:46-54.
- [Google Scholar]
- Proliferative glomerulonephritis with monoclonal IgG deposits secondary to chronic lymphocytic leukemia. Report of two cases. Nephrol Dial Transplant. 2011;26:2712-4.
- [Google Scholar]
- Proliferative glomerulonephritis with monoclonal IgG deposits recurs in the allograft. Clin J Am Soc Nephrol. 2011;6:122-32.
- [Google Scholar]
- Proliferative glomerulonephritis with monoclonal IgG deposits recurs or may develop de novo in kidney allografts. Am J Kidney Dis. 2011;58:276-81.
- [Google Scholar]
- Recurrent proliferative glomerulonephritis with monoclonal IgG deposits of IgG2λ subtype in a transplanted kidney: A case report. Am J Kidney Dis. 2013;62:587-90.
- [Google Scholar]
- A case of recurrent proliferative glomerulonephritis with monoclonal IgG deposits after kidney transplant treated with plasmapheresis. Case Rep Nephrol Urol. 2012;2:46-52.
- [Google Scholar]
- Aggregation of gamma-G3 proteins: Relevance to the hyperviscosity syndrome. J Clin Invest. 1970;49:610-21.
- [Google Scholar]




